A Convenient Method to Prepare Labile FMN Derivatives

Department of Biochemistry and Molecular Biology, The University of Georgia, Athens, Georgia 30602, USA
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(10), 825-830; https://doi.org/10.3390/61000825
Submission received: 30 September 2001
/
Accepted: 10 October 2001
/
Published: 31 October 2001
{kind=link}
Abstract
:A simple method for selective phosphorylation of ribityl containing starting materials to afford FMN derivatives is presented. The method is in particular valuable for the synthesis of labile FMN derivatives.
Introduction
Of the available published procedures usually used to phosphorylate riboflavin and its derivatives to synthesize FMN and FMN analogues, none yield a fairly homogeneous product with respect to the ribityl side chain phosphorylation. The most commonly used procedure is that developed by Flexner and Farkas [1], using phosphorus oxychloride. Although this synthetic approach has been improved by Scola-Nagelschneider and Hemmerich [2], the resulting FMN still contained a considerable amount of by-products. Commercial FMN, usually prepared by the same methods, contains only about 70% of FMN, the residual 30% being made up of various mono- and diphosphates [3]. Although biochemical methods are now available for selective phosphorylation of riboflavin and its derivatives, these methods are currently only suited to produce relatively small amounts. We have therefore developed a new procedure allowing the preparation of larger amounts by a simple method followed by a facile purification, yielding a rather homogeneous product. The method is in particular very suitable for the preparation of labile (oxygen- or light-sensitive) FMN derivatives. In addition the method can also be helpful in the phosphorylation of riboflavin analogues substituted with bulky groups at the 8-α position which are not substrates for flavokinase, to form the corresponding FMN derivatives.
Results and Discussion
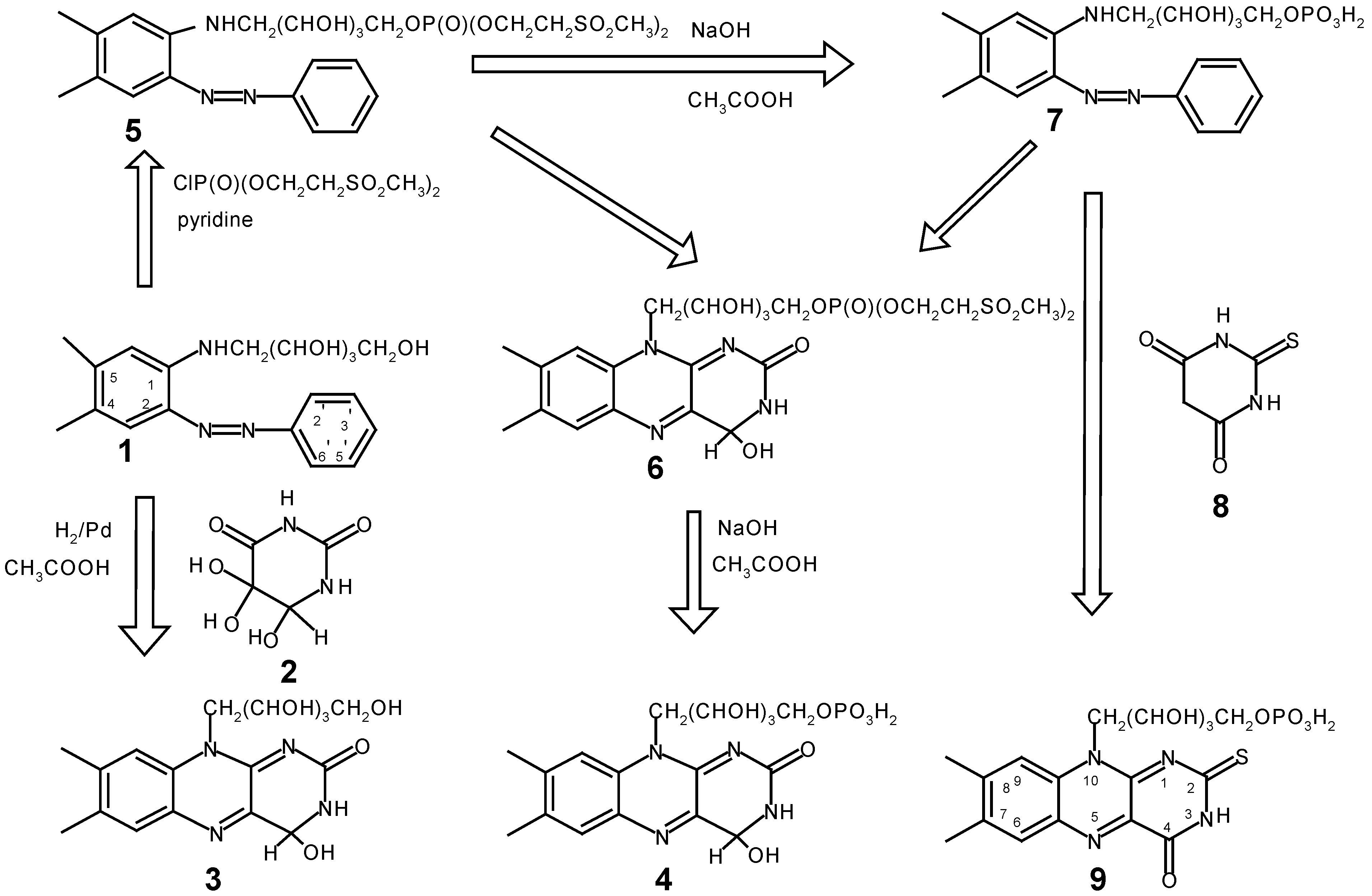
The synthesis of the oxygen-sensitive 2-thio-FMN has been described by Föry and Hemmerich [4]. The procedure is very tedious because of the time-consuming work-up methods needed to remove the oxidation product (FMN) generated during the synthesis and purification. Although 3,4-dihydro-riboflavin (Compound 1, Scheme 1) is easily synthesized, phosphorylation by conventional methods would lead to FMN which it is difficult to remove from the reaction mixture. We now present a new method yielding selectively phosphorylated products in pure form, involving only a few synthetic steps. Beld et al [5] have synthesized a new phosphorylating agent, bis[2-(methylsulfonyl)ethyl] phosphochloridate, which is easily synthesized in almost quantitative yield and exhibits a higher specificity with regard to the phosphorylation site of nucleosides than other agents. Compound 1 (Scheme 1), a versatile chemical for the synthesis of many riboflavin derivatives, was phosphorylated with bis[2-(methylsulfonyl)ethyl] phosphochloridate in pyridine abs. at room temperature to give 5 in a fair yield. It has been noticed that exceeding the reaction time of 3.5 h leads to the formation of an increasing amount of an unidentified by-product with time. The course of the reaction can easily be monitored by tlc.
Scheme 1.

The work-up procedure includes extraction of the product, dissolution in phosphate buffer and extraction with n-butanol, followed by column chromatography and evaporation of the solvent to give a crystalline product. The pure compound is hygroscopic and should be stored under dry conditions in a refrigerator, because some secondary product(s) are formed with time. This observation has not been further pursued but could be ascribed to the reaction of some residual MeOH contained in the final product (see [5] for an explanation). It should also be noted that all NMR spectra showed the presence of both syn and anti configurations.
In principle 5 could be condensed with 2 or 8 to give the corresponding flavin derivatives which then must be deprotected to yield the desired FMN compounds. Compound 6 was synthesized in good yield but the conversion to 4 led to a considerable amount of FMN. Therefore both compounds were synthesized from 7. The conversion of 5 to 7 in 5N NaOH occurred in high yield. Since 7 was pure as judged by tlc, no attempts were undertaken to obtain it in crystalline state, because its direct use did not jeopardize the synthesis of 4 and 9 in any way. Compound 7 can be used directly in the condensation with 8 to afford 2-thio-FMN, whereas alloxan derivatives require ortho-diaminobenzene derivatives [6]. Thus 7 was reduced with H2/Pd/C at atmospheric pressure and the reaction product used in situ in the condensation with 2 to afford 4.
Conclusions
A facile method to synthesize labile FMN derivatives with a high degree of selective phosphorylation is presented. The method is also valuable for phosphorylation of riboflavin derivatives containing bulky groups not accepted by kinase as substrates.
Experimental
General
1H- and 13C-NMR spectra were recorded on a Bruker CXP 300 spectrometer. The chemical shifts are reported in ppm relative to TMS (1H, 13C) or ext. 85% H3PO4 (31P). All reagents and chemicals were obtained from Aldrich Chemical Company (USA) or Eastman Organic Chemicals (USA) and were used as received. 4,5-Dimethyl-2-phenylazo-N-ribityl-aniline (1) was a generous gift of Hoffmann‑La Roche & Co. Ltd, Basle, Switzerland. Isodialuric acid (2) was prepared from 5‑aminouracil according to the procedure of Behrend and Roosen [7]: 13C-NMR (CD3OD): 169.3, C(6); 154.1, C(2); 93.3, C(5); 77.7, C(4); 1H-NMR (D2O): 4.63, C(4)H. The phosphorylating agent bis[2-(methylsulfonyl)ethyl] phosphochloridate was synthesized by the method of Beld et al. [5].
4,5-dimethyl-2-phenylazo-N-ribityl 5'phosphate-bis[2-(methylsulfonyl)ethyl]-aniline (5)
Compound 1 (5.0 g) was dissolved in absolute pyridine (100 mL) and the phosphorylating agent (7.5 g) was added. The clear solution was then stirred at room temperature. After one hour about 50% of the starting material was converted to the product as evidenced by tlc (Silica Gel Plastic Plates, Kodak no. 13181, mobile phase: n-butanol/EtOH/H20=50/15/35). At this point additional phosphorylating agent (3.0 g) was added. After a total reaction time of 3.5 h the reaction was almost completed. It was noted that by-product(s) formed after longer reaction times, therefore the reaction was quenched after 3.5 h by pouring the reaction mixture into 1 M pH 7 phosphate buffer (600 mL), containing ice. The small amount of precipitate formed, some residual starting material, was removed by filtration and the clear solution several times extracted with n-butanol. The collected organic phase was then evaporated to dryness under reduced pressure. The residue was dissolved in ethyl acetate and chromatographed on a Kieselgel 60 column (7 x 15 cm). Elution with ethyl acetate gave a small amount of an unidentified by-product and residual starting material together with a small amount of the aforementioned secondary product. The main product was eluted with ethyl acetate-10% methanol. The solution of the main fraction was then evaporated to dryness under reduced pressure and the residue dissolved in ethyl acetate, and again freed from solvent by evaporation. This procedure was repeated until a crystalline product was obtained. The yield of 5 was 6.5 g (72 %). The product should be stored under dry conditions (hygroscopic!). 1H‑NMR (DMSO): 8.95 (1 H, s, broad), NH; 7.81 (1 H), 7.75 (1 H), 7.25 (1 H), 7.50 (1 H), 7.47 (1 H), 7.41 (1 H, d), 6.72 (1 H, d), aromatic protons; 5.7‑5.0 (7 H, multiplet), ribityl side-chain; 4.01 (2 H, doublet), 3.62 (2 H, doublet), O-CH2-CH2-SO2; 3.00 (3 H), 2.97 (3 H), SO2CH3; 2.31 (2H), NH-CH2; 2.25 (3 H, s), CH3 (5α); 2.20 (3 H, s), CH3 (4α). 13C-NMR (DMSO; proton-decoupled): 156.2 (s), C(2); 146.7 (s), C(1), 146.2 (s), C(1’); 138.2 (s), C(4); 133.4 (d), C(4’); 133.1 (d), C(2’,6’); 126.9 (s), C(5 ); 125.4 (d), C(3’,5’); 117.1 (s), C(3); 117.0 (s), C(6); 76.6 (d), CH2SO2*; 74.1 (d), OCH2*; 70.1 (d), 1ε*; 67.0 (d), 1δ ; 62.5 (d), 1γ*; 59.0 (d), 1ζ*; 49.3 (d), SO2CH3; 48.6 (d), SO2CH3; 46.1 (d), NHCH2; 24.1 (s), CH3 C(5α); 22.1 (s), CH3 C(4α); (Note: * indicates tentative assignments). 31P-NMR: 2.6 (d); 2.7 (d); syn and anti forms.
7,8-dimethyl-10-ribityl-5'phosphate-bis(2-(methylsulfonyl)ethyl)-3,4-dihydro-isoalloxazine (6)
Compound 5 (0.5 g) was suspended in glacial acetic acid (15 mL) and dissolved by warming, then Pd/C was added and the compound hydrogenated under atmospheric pressure. After completion of the hydrogenation reaction the colorless solution was filtered under anaerobic conditions into a 250 mL round bottom flask and the solvent evaporated under reduced pressure to a small volume of about 3-4 mL. The residual solution was then flushed with nitrogen while isodialuric acid (2, 0.3 g), dissolved in warm MeOH (3mL) was added. To the mixture was then added ethyl acetate (25 mL) and the mixture flushed with nitrogen to provide an anaerobic solution. The solution was sealed with a rubber stopper and the reaction mixture stirred over night at room temperature and protected from light. The precipitate formed was filtered, washed with ethyl acetate and dried with ether. The yield of crude product was 0.3 g (60 % of theory). The crude product was purified by dissolution in warm MeOH, filtration from insoluble material and the clear solution diluted with ethyl acetate until a slight turbidity appeared. This solution was stored in a refrigerator over night. The pale yellow material formed was filtered, washed with ethyl acetate and dried with ether. The yield was 0.2 g of pure 6 (40 %) as estimated by tlc The product showed the expected UV-visible and fluorescence properties [8].
3,4-Dihydro-riboflavin (3):
It was prepared in the same way as 6. Starting with 1.8 g of 1 and 1.1 g of 2, 1.6 g of crude product was obtained which was purified by dissolution in warm MeOH, filtration, followed by chromatography on Kieselgel 100 ( Merck). The desired product was eluted with MeOH and the eluate evaporated to dryness. The yield of pure 3 was 0.6 g (33 %). 13C-NMR (CD3OD): 163.48, C(2); 151.67, C(8); 145.75, C(10a); 142.21, C(7); 137.89, C(5a); 136.57, C(4a); 133.43, C(6); 132.26, C(9a); 119.51, C(9); 85.05, C(4); 76.52, C(10δ); 75.45, C(10γ); 72.92, C(10β); 66.13, C(10ε); 48.00, C(10α); 22.01, C(8α); 20.49, C(7α).
4,5-Dimethyl-2-phenylazo-N-ribityl 5'-phosphate-aniline (7)
Compound 5 (1.6 g) was dissolved in a mixture of dioxane/MeOH (3:1, 20 mL) and 5N NaOH (4 mL) was added. After 3 min the reaction was quenched by the addition of glacial acetic acid (5 mL) and the reaction mixture concentrated to a small volume under reduced pressure. To this solution 0.1M pH 7 phosphate buffer (100mL), was added and the mixture extracted several times with n-BuOH. The organic phase was then evaporated to dryness under reduced pressure, yielding a semi-crystalline product which was not further purified because it is very difficult to crystallize due to its highly hygroscopic nature. However it was shown to be almost pure by tlc. The yield of 7 was 1 g (93 %; the actual yield must be lower since it was not possible to completely free the product from residual solvents but these did not affect the use of 7 in further synthetic work).
3,4-Dihydro-FMN (4)
Compound 7 (0.5 g) was dissolved in glacial acetic acid (30 mL) with slight warming and then reduced with hydrogen in the presence of Pd/C at atmospheric pressure. The colorless solution was then filtered under anaerobic conditions into a round bottom flask and the solvent reduced to a small volume under reduced pressure. To this solution, which was kept under a stream of nitrogen, was added isodialuric acid (300 mg) dissolved in warm MeOH (5 mL) which was also flushed with nitrogen. To this solution ethyl acetate was then added until a slight turbidity formed. The solution was further flushed with nitrogen for about 10 min, the flask then sealed with a rubber stopper and the solution allowed to stand overnight at 4°C. The precipitate was filtered and washed with ether. Fluorescence spectra and tlc demonstrated the absence of FMN in 4. The yield of pure 4 was 250 mg (48 %).
2-Thio-FMN (9)
Compound 7 (0.5 g) was dissolved in glacial acetic acid (30 mL) in a three-necked flask equipped with a reflux condenser, thermometer and a N2-inlet. The content was warmed to 50°C, thiobarbituric acid (8, 300 mg) added and the reaction mixture stirred overnight at 50°C under a nitrogen atmosphere. The dark red precipitate was filtered and washed with ether. Yield: 250 mg. A second crop of 100 mg was obtained from the mother liquor by addition of an equal volume of ethyl acetate. Since the product did not contain any FMN nor 7 as evidenced by tlc and fluorescence spectra, the product was considered as pure. Total yield of 9 was 350 mg (92 %).
Acknowledgements
We thank Dr. J. Vervoort for recording some of the NMR spectra.
References
- Flexner, L. A.; Farkas, W. G. Preparation of Riboflavin Phosphate Intermediates. U.S. Patent 1952, 2,610,176, [Chem. Abstr.1953, 47,8781a]. [Google Scholar]
- Scola-Nagelschneider, G; Hemmerich, P. Synthesis, Separation, Identification and Interconversion of Riboflavin Phosphates and Their Acetyl Derivatives: A Reinvestigation. Eur. J. Biochem. 1976, 66, 567–577. [Google Scholar]
- Nielsen, P.; Rauschenbach, P.; Bacher, A. Phosphates of Riboflavin and Riboflavin Analogs: A Reinvestigation by High-Performance Liquid Chromatography. Anal. Biochem. 1983, 130, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Föry, W.; Hemmerich, P. Synthese Funktionell Abgewandelter Flavokoenzyme: 3-Alkyl und 2-Alkyliminoflavin-mononucleotide and Flavin-adenin-dinucleotide. Helv. Chim. Acta 1967, 50, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Beld, A.; Claesen, C. A. A.; Roersma, E. S.; Schippers, W. J. M.; Keizer, L. M.; Tesser, G. I. Bis[2-(methylsulfonyl)methyl] phosphochloridate, a New Phosphorylating Agent. Recl. Trav. Chim. Pays-Bas 1984, 103, 196–202. [Google Scholar] [CrossRef]
- Müller, F. Chemistry and Biochemistry of Flavoenzymes; Müller, F., Ed.; CRC Press: Boca Raton, FL, 1991; Vol. 1, Chapter 1; p. 4. [Google Scholar]
- Behrend, R.; Rosen, O. Synthese der Harnsäure. Justus Liebigs Ann. Chem. 1889, 251, 235–256. [Google Scholar] [CrossRef]
- Müller, F.; Massey, V.; Heizmann, C.; Hemmerich, P.; Lhoste, J. M.; Gould, D. C. The Reduction of Flavins by Borohydride: 3,4-Dihydroflavins. Structure, Absorption and Luminescence. Eur. J. Biochem. 1969, 9, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples are available from MDPI.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Müller, F.; Lee, J. A Convenient Method to Prepare Labile FMN Derivatives. Molecules 2001, 6, 825-830. https://doi.org/10.3390/61000825
AMA Style
Müller F, Lee J. A Convenient Method to Prepare Labile FMN Derivatives. Molecules. 2001; 6(10):825-830. https://doi.org/10.3390/61000825
Chicago/Turabian StyleMüller, Franz, and John Lee. 2001. "A Convenient Method to Prepare Labile FMN Derivatives" Molecules 6, no. 10: 825-830. https://doi.org/10.3390/61000825