Results and Discussion
Quinazoline-4-thiones can be synthesised by several ways: e.g. it is possible to transform the corresponding oxygen derivative into its sulphur analogue by treating it with phosphorus pentasulphide [
4] or Lawesson’s reagent [
5]. It is also possible to make use of the reactivity of suitably substituted isothiocyanates [
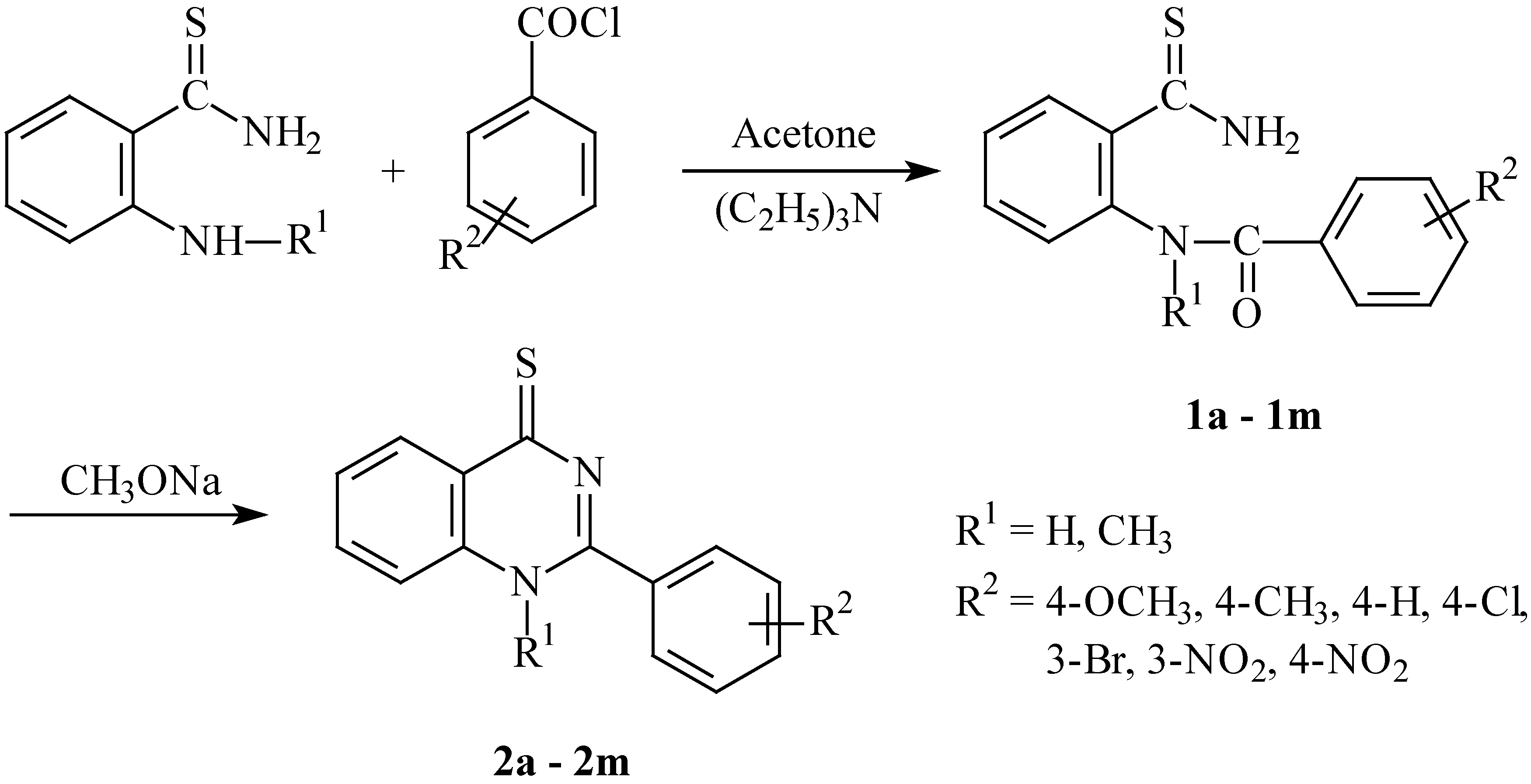
6]. In our work we have chosen the way of constructing the heterocyclic skeleton which consists in the acylation of 2-aminothiobenzamide or 2-methylamino-thiobenzamide with substituted benzoyl chlorides and subsequent ring closure of the thus obtained 2-benzoylaminothiobenzamides (
1a-
m) in basic medium to give the quinazoline-4-thiones (
2a-m) (
Scheme 1).
Scheme 1.
- Preparation of 2-benzoylaminothiobenzamides and 2-phenylquinazoline-4-thiones
Scheme 1.
- Preparation of 2-benzoylaminothiobenzamides and 2-phenylquinazoline-4-thiones
The starting 2-aminothiobenzamide and 2-methylaminothiobenzamide were prepared by two-step syntheses from 2-aminobenzamide and 2-methylaminobenzamide, respectively. In the first step we prepared the corresponding pyridinium salt by treatment with phosphorus pentasulphide [
7], and in the second step the pyridinium salt was transformed into the respective thioamide by our original hydrolysis method in a toluene-water system. The benzoylation of the thioamides thus obtained was carried out by treatment with commercial benzoyl chlorides in acetone solvent. The reaction gives 2-benzoylaminothiobenzamides
1a-
m, which are very sensitive to both basic and acidic media as well as to air oxygen and heating (the melting temperature intervals are relatively broad, which is a consequence of the decomposition during m. p. determination). Due to its low stability it was impossible to prepare compound
1e (R
2 = 4-NO
2) with sufficient purity for characterisation. In the syntheses any contact with metals must be avoided, otherwise the
1H-NMR spectra show broadened signals due to the presence of paramagnetic substances – most probably metal complexes of the thioamides. The ring closure of 2-benzoylaminothiobenzamides
1a-
m to the corresponding 2-(subst. phenyl)quinazoline-4-thiones
2a-
m is easily achieved in solutions of sodium methoxide even at room temperature (
Scheme 1).
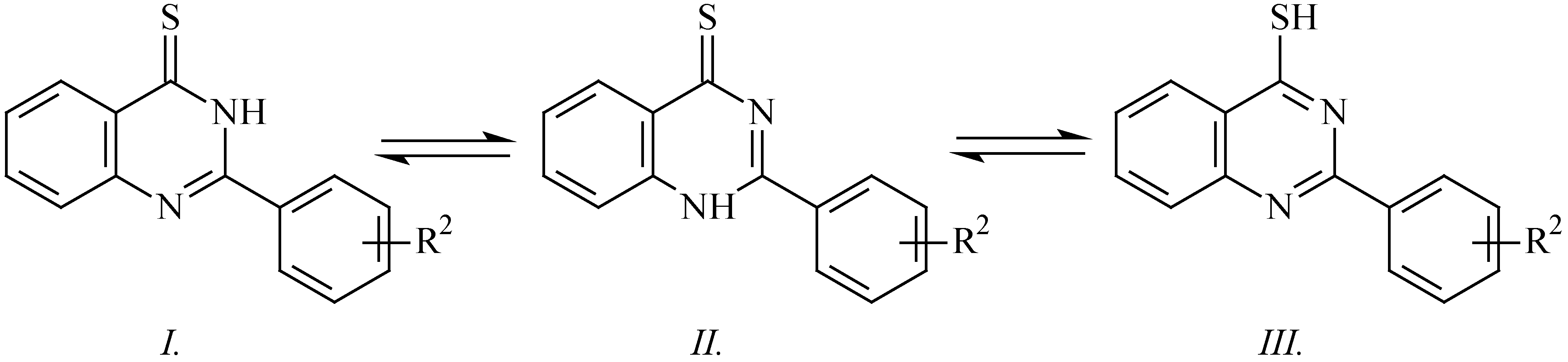
The newly synthesised 2-(subst.phenyl)quinazoline-4-thiones
2a-
f can exist in three tautomeric forms (
Scheme 2) similar to those of the parent compound, the unsubstituted quinazoline-4-thione itself, whose structure was discussed previously, but only on the basis of its UV [
8] and IR spectra [
9]. Therefore, we have focused our attention on interpretation of
13C-NMR, IR, Raman and UV spectra. The IR spectra of the substances mentioned exhibit a broad band in the interval of 3150 – 3300 cm
–1, which indicates the presence of NH group (valence vibrations). This finding together with the fact that the Raman spectrum does not show any band in the region of 2500 – 2600 cm
–1 (the valence vibration of SH group) exclude structure
III. Also the chemical shift of C4 in the region of 187.9 – 188.2 ppm is much higher than that of 2-aryl-4-allylthioquinazolines (157.8 – 158.6 ppm) [
10]. The differentiation between structures
I and
II is possible on the basis of comparison of the UV spectra, or better still, that of the
13C-NMR shifts of C=S group. The absorption maximum of derivatives
2a-
f is at 360 nm, while that of the derivatives
2g-
m (whose methyl group prefers a tautomeric arrangement close to structure
II) is at 388 nm. A similar conclusion follows from the
13C-NMR shifts of C=S group: those of derivatives
2a-
f lie in the region of 187.9 – 188.2 ppm, that of 2-phenyl-3-methylquinazoline-4-thione is at 189.2 ppm, and those of derivatives
2g-
m lie in the region of 198.7 – 199.0 ppm. The shifts mentioned indicate that the preferred tautomeric form is
I as it is the case with the analogous 2-phenylquinazolin-4-ones too.
Scheme 2.
- Possible tautomeric structures of 2-(subst.phenyl)quinazoline-4-thiones
Scheme 2.
- Possible tautomeric structures of 2-(subst.phenyl)quinazoline-4-thiones
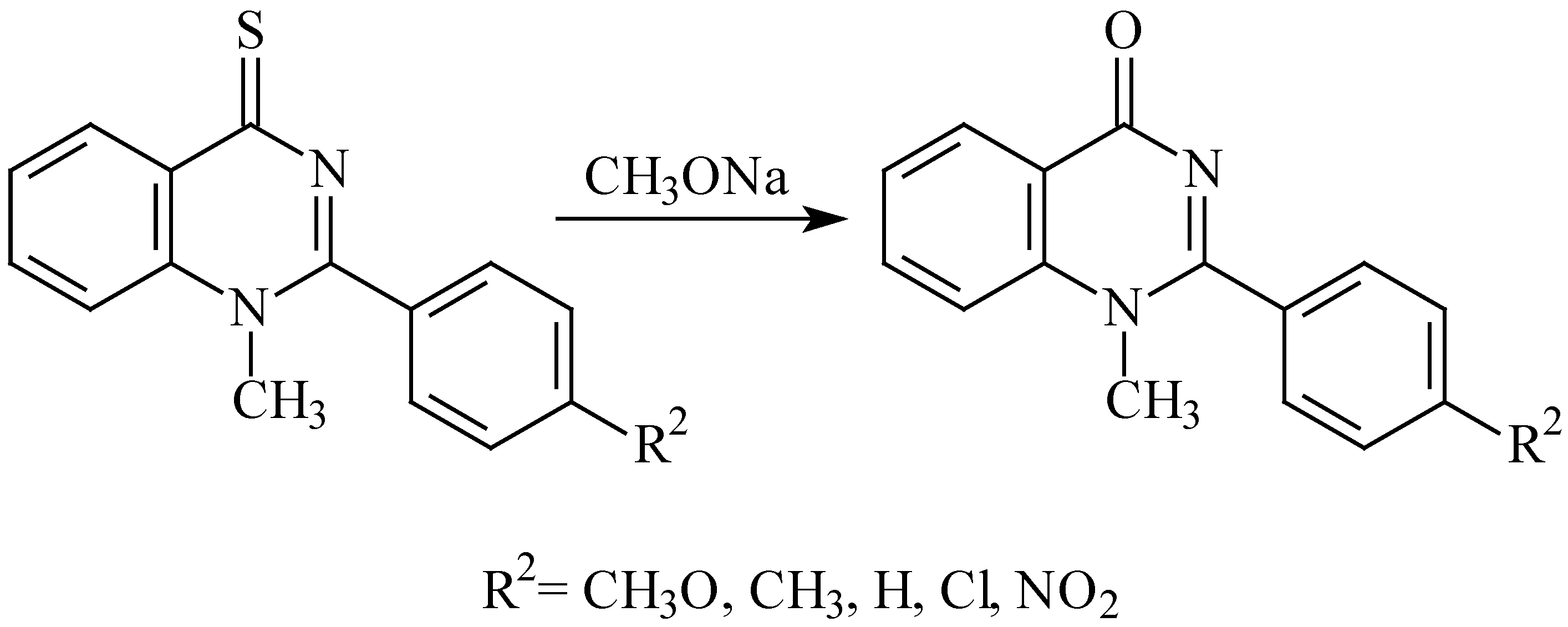
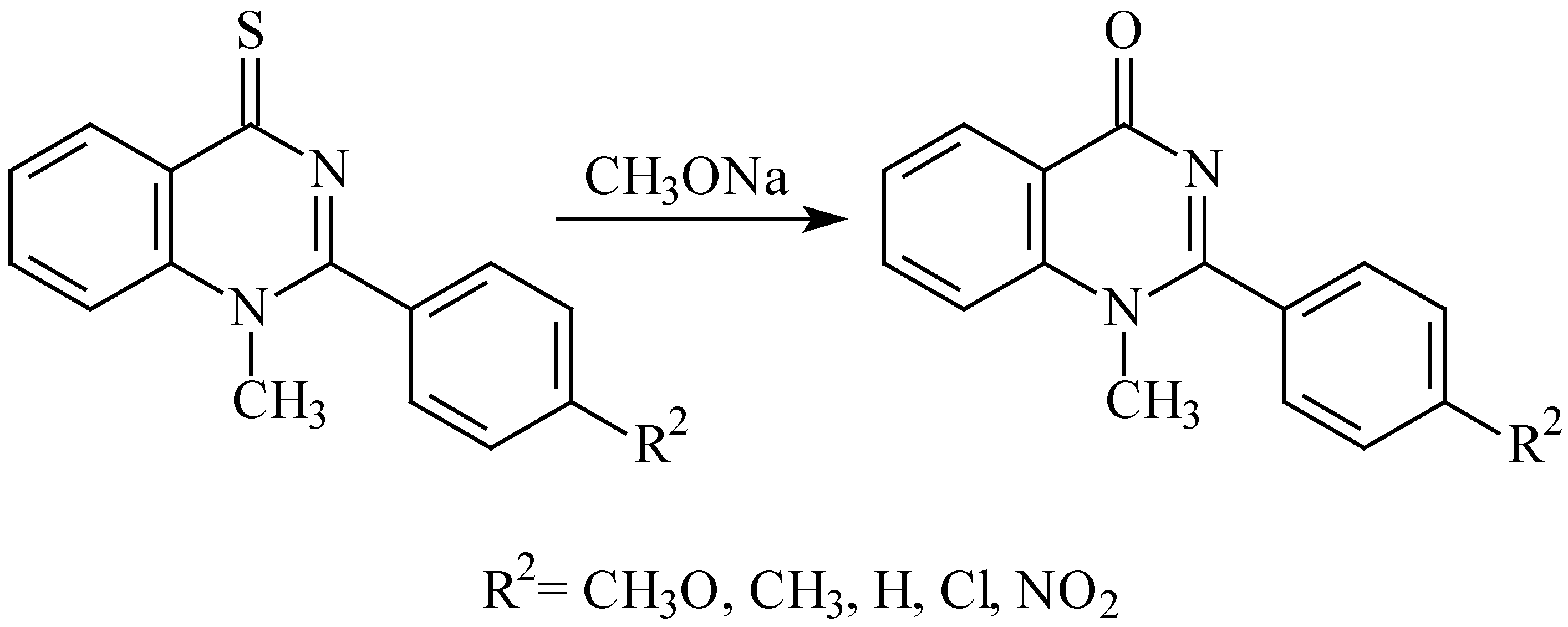
When synthesising the
N-methyl derivatives
2g-
m we observed that the cyclisation is followed by desulphuration giving the corresponding 2-phenylquinazolin-4-one derivatives. This finding was verified on preparative scale: we refluxed 1-methyl-2-(4-chlorophenyl)quinazoline-4-thione (
2j) in sodium methoxide and obtained 1-methyl-2-(4-chlorophenyl)quinazolin-4-one in 67% yield (
Scheme 3). Similar desulphurations of quinazoline-4-thiones to quinazolin-4-ones were described earlier but the reaction conditions were substantially different: either a preliminary
S-alkylation [
11,
12,
13] or treatment with hydrogen peroxide [
13].
Scheme 3.
- Desulphuration of 1-methyl-2-(subst.phenyl)quinazoline-4-thiones
Scheme 3.
- Desulphuration of 1-methyl-2-(subst.phenyl)quinazoline-4-thiones
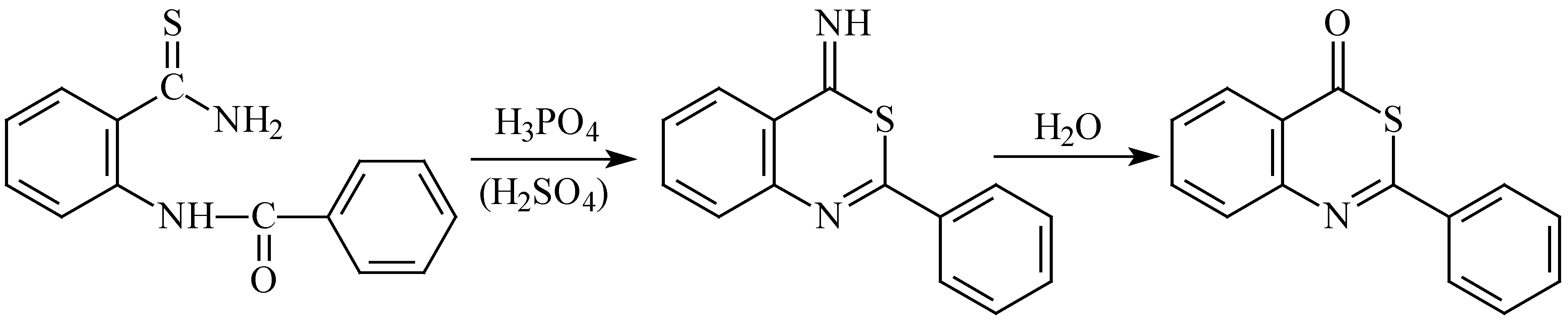
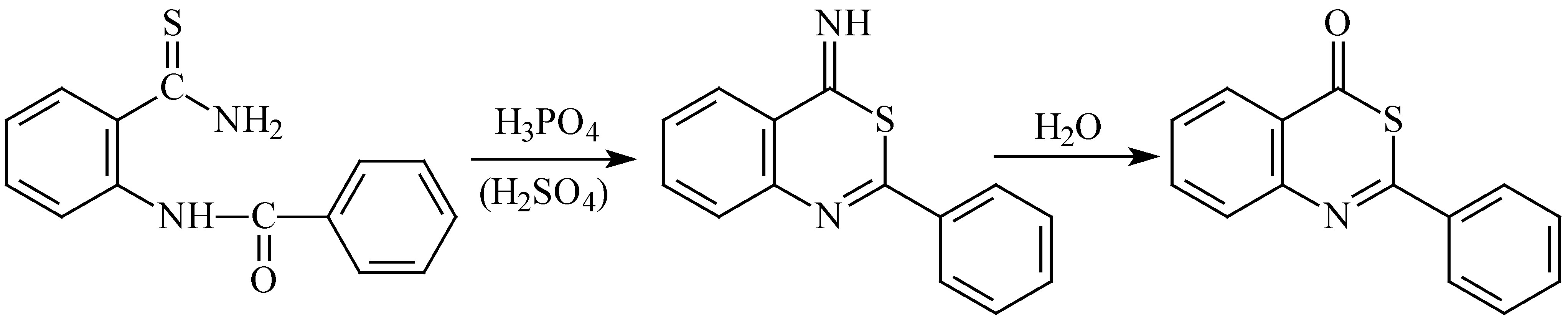
Moreover, we have found that in strong acid medium 2-benzoylaminothiobenzamide (
1a) undergoes ring closure through its sulphur atom, and the same reaction can also be presumed with the other derivatives (
1b-
f). A similar reaction was described earlier with thioureide esters [
14] and selenoureide esters [
15]. This finding can be interpreted by the SH group being a stronger nucleophile in acid medium than the protonated nitrogen of thioamide group, the opposite being true in basic media. When compound
1a was stirred in concentrated sulphuric acid at room temperature for three days, the products isolated from the resulting reaction mixture involved (besides the unreacted starting substance
1a) two cyclisation products: 2-phenyl-4-iminobenzo[
d-1,3]thiazine and 2-phenylbenzo[
d-1,3]thiazin-4-one, the latter being formed by subsequent hydrolysis during isolation (
Scheme 4).
Scheme 4.
- Cyclisation of 2-benzoylaminothiobenzamide (1a) in acid medium
Scheme 4.
- Cyclisation of 2-benzoylaminothiobenzamide (1a) in acid medium
After prolonging the reaction time (to 7 days) and increasing of reaction temperature (60 °C) the reaction products again included the starting material 1a, and a new product also appeared: bis(2-phenylquinazolin-4-yl)sulphide formed by a parallel reaction. In order to suppress the oxidation, the ring closure was carried out in anhydrous phosphoric acid at room temperature (14 days), which was followed by hydrolysis in 50% aqueous methanol (30 min reflux). The HPLC-MS analysis of the product showed that it contained 15% starting thioamide 1a and 85% 2-phenylbenzo[d-1,3]thiazin-4-one; the latter compound was then obtained in pure state by recrystallisation from aqueous methanol (yield 49%).
The structure of the 2-benzoylaminothiobenzamides (1a-m) and cyclisation products (2a-m) was verified by spectral methods (1H- and 13C-NMR, IR) and elemental analysis.
Experimental
General
The 1H- and 13C-NMR spectra were measured at 360.14 and 90.57 MHz, respectively, at 25 °C, using a Bruker AMX spectrometer. Compounds 1a-m and 2a-m were measured as saturated solutions in hexadeuteriodimethyl sulphoxide (DMSO-d6), and the chemical shifts are referenced to tetramethylsilane (δ(1H) = 0) and the solvent signal (δ(13C) = 39.6). The CH, CH3, and Cquart groups in 13C-NMR spectrum were differentiated by means of the APT method.
The IR spectra were measured in Nujol using a Perkin–Elmer 684 apparatus and a KBr cell. The Raman spectra were measured in dimethyl sulphoxide using a Bruker IFS 55 apparatus with an FRA 106 extension. For the source we used the Nd:YAG laser with the excitation frequency of 9394 cm–1.
The mass spectra were measured with a VG Platform II mass spectrometer (Micromass, Manchester, UK) using chemical ionisation at atmospheric pressure (APCI) and a quadrupole analyser (0-3000 Da). Before the mass spectrometer there was inserted a separation apparatus consisting of a Waters 616 high-pressure pump, Waters 717 autosampler and a Waters 966 UV detector (all from Waters, Milford, USA), and a Separon SGX C18 octadecyl silica glass cartridge column (150 × 3 mm i.d., 7 μm particle size; Tessek Ltd., Prague, Czech Republic). A mixture of 70% acetonitrile and 30% redistilled water was used as the mobile phase. The effluent from the liquid chromatography was directly introduced into a quadrupole mass spectrometer equipped with APCI probe operated in positive ion mode. The data was acquired in the m/z range 15-600 at 1.9 s per scan. In the APCI mode, the temperature of the ion source and of the probe were 100 and 500°C, respectively. The cone voltage was set at 10 V for positive-ion APCl.
2-Aminothiobenzamide
A 250-mL flask was charged with 2-aminobenzamide (13.6g, 0.1 mol), phosphorus penta-sulphide (22.3g, 0.1 mol) and pyridine (70 mL). The mixture was refluxed 1.5 h, whereupon it was cooled and poured onto ice water (250 mL). The separated yellow crystals of the pyridinium salt of 2-mercapto-2-thioxo-2,3-dihydro-1H-2λ5-benzo[1,3,2]diazaphophinine-4-thione were collected by filtration and recrystallised from a mixture of water and dimethylformamide (5:1). Yield 20.8g (64%) yellow crystals, m.p. 190-201 °C.
A 500-mL flask was charged with the pyridinium salt of 2-mercapto-2-thioxo-2,3-dihydro-1
H- 2λ
5-benzo[1,3,2]diazaphophinine-4-thione (19.5g, 0.06 mol) along with water (70 mL), toluene (70 mL), and conc. hydrochloric acid (1.3 mL). The reaction mixture was refluxed until the solid portion completely dissolved and then for another 4 h, whereupon it was cooled to room temperature, and the separated crystals were collected by filtration and recrystallised from water. Yield: 5g (54.6%) yellow crystals, m.p. 118-121 °C (in agreement with ref. [
16]).
2-Methylaminothiobenzamide
This substance was prepared in the same way as 2-aminothiobenzamide. The first step gave 23.5g (69%) of the pyridinium salt of 1-methyl-2-mercapto-2-thioxo-2,3-dihydro-1
H-2λ
5-benzo[1,3,2]diaza-phophinine-4-thione, m.p. 165-170 °C. The second step (hydrolysis of this salt) gave 7.9g (79.1%) 2-methylaminothiobenzamide, m.p. 114-115 °C (in agreement with ref. [
17]).
2-Amino-N-methylthiobenzamide
This substance was prepared in the same way as 2-aminothiobenzamide. The first step gave 20g (59%) of the pyridinium salt of 3-methyl-2-mercapto-2-thioxo-2,3-dihydro-1
H-2λ
5-benzo[1,3,2]-diazaphophinine-4-thione, m.p. 145-150 °C (ref. [
7] gives m.p. 168-169 °C). The second step (hydrolysis of this salt) gave 6.3g (64.3%) 2-amino-
N-methylthiobenzamide, m.p. 96-98 °C (in agreement with ref. [
18]).
2-Benzoylaminothiobenzamides 1a-m
A 100-mL three-necked flask equipped with a magnetic stirrer, dropping funnel, and argon inlet was charged with the appropriate thioamide (10 mmol), dry acetone (40 mL), and triethylamine (10 mmol). The solution formed was treated with the corresponding benzoyl chloride (10 mmol) dissolved in acetone (10 mL), added within ca. 5 min. The reaction mixture was stirred under an argon atmosphere at room temperature 0.5-1 h. The separated triethylamine hydrochloride was collected by filtration on a pressure filter (sintered glass), and the filtrate was evaporated in vacuum at room temperature. The evaporation residue was mixed with ice-cold methanol (10 mL). The separated crystals were collected under argon and carefully washed with ice-cold methanol (3×10 mL). If the product thus obtained did not possess the required quality, it was recrystallised from methanol at room temperature (max. 25 °C). The yields, melting points, and
1H- and
13C-NMR spectra are summarised in
Table 1,
Table 2,
Table 3,
Table 4 and
Table 5.
2-Phenylquinazoline-4-thiones 2a-m
A 100-mL flask was charged with the appropriate 2-benzoylaminothiobenzamide
1a-
m (2 mmol) and ethanol (50 mL). The solution formed was treated with 1M sodium methoxide (1 mL), and the reaction mixture was refluxed 1 h, whereupon it was neutralised with acetic acid to pH ~ 7 and concentrated to crystallisation. The crystalline product obtained on cooling was recrystallised from toluene, methanol, or dimethylformamide. The yields, melting points,
1H- and
13C-NMR spectra are summarised in
Table 6,
Table 7,
Table 8,
Table 9 and
Table 10.
2-Benzoylamino-N-methylthiobenzamide
This substance was prepared in the same way as the 2-benzoylaminothiobenzamides 1a-m. The yield of the product thus obtained was 1.7g (62%), m. p. 192-193 °C. 1H-NMR (δ): 3.18 (s, 3H, CH3), 7.28 (m, 1H, H-3), 7.41 (m, 1H, H-5), 7.53 (m, 1H, H-4), 7.58 – 7.68 (m, 3H, H-m + H-p), 7.95 (m, 2H, H-o), 8.20 (m, 1H, H-2), 10.78 a 10.86 (2×bs, 2H, NH).
2-Phenyl-3-methylquinazoline-4-thione
The preparation was the same as that used for 2-phenylquinazoline-4-thiones 2a-m. Yield 0.4g (80%), m. p. 134-136 °C. 1H-NMR (δ): 3.85 (s, 3H, CH3), 7.60 – 7.64 (m, 3H, H-m +H-p), 7.67 (m, 1H, H-3), 7.75-7.79 (m, 3H, H-o + H-5), 7.94 (m, 1H, H-4) 8.74 (m, 1H, H-2). 13C-NMR (δ): 43.8 (CH3), 128.9 (C-4a), 128.9 (C-8), 129.2 (C-m), 129.3 (C-6), 129.4 (C-o), 130.8 (C-5), 131.0 (C-p), 135.6 (C-7), 136.7 (C-i), 143.0 (C-8a), 156.0 (C-2), 188.9 (C=S).
Ring Closure of 1a in Sulphuric Acid
A 50-mL flask was charged with 2-benzoylaminothiobenzamide (1a, 0.5 g, 1.95 mmol) together with 96% sulphuric acid (5 mL) and acetic anhydride (3 mL). The mixture was stirred at room temperature 3 days, whereupon it was poured onto crushed ice (50g). The separated crystals were collected by suction, washed with water, with 10% aqueous sodium bicarbonate (30 mL), and again with water. One major and two minor components were identified by means of HPLC-MS. The minority components were identified as: (1) the starting material, 2-benzoylaminothiobenzamide (1a), MS (m/z,%): [M+H]+ - (257.1, 93); [M–H2S+H]+ - (223.1, 100) and (2) 2-phenyl-4-imino-benzo[d-1,3] MS (m/z,%): [M+H]+ - (239.1, 100); [M–H2S+H]+ - (205.0, 12). The major component was 2-phenylbenzo[d-1,3]thiazin-4-one MS (m/z): [M+H]+ - (240.1).
Ring Closure of 1a in Anhydrous Phosphoric Acid
A 50-mL flask was charged with 2-benzoylaminothiobenzamide (
1a, 0.5 g, 1.95 mmol) together with anhydrous phosphoric acid (10 mL, 85% H
3PO
4 + 6.7 g P
2O
5). The mixture was stirred at room temperature 14 days. Then it was poured onto crushed ice (50g) and the separated crystalline solid was collected by suction, washed with water, with 10% aqueous sodium bicarbonate (30 mL), and again with water. The filter cake was suspended in 50% aqueous methanol (60 mL) and refluxed 30 min. The crystalline solid separated on cooling was collected by suction and recrystallised from methanol with addition of charcoal. Yield 0.23g (49%) long yellowish needles, m.p. 113-116 °C (ref. [
19] gives m.p. 116 °C). MS (m/z): [M+H]
+ - 240.1.
Table 1.
– Melting points and elemental analyses of the 2-(3 and 4-subst. benzoylamino)-thiobenzamides.
Table 1.
– Melting points and elemental analyses of the 2-(3 and 4-subst. benzoylamino)-thiobenzamides.
| Compound | R1 | R2 | m.p.* (°C) Yield (%) | Formula M.W. | Elemental analysis Calculated/Found (%) |
|---|
| C | H | N | S | X |
|---|
| 1a | H | H | 182-184 | C14H12N2OS | 65.60 | 4.72 | 10.93 | 12.51 | |
| 48 | 256.32 | 65.48 | 4.79 | 10.89 | 12.59 |
| 1b | H | 4-CH3 | 205-210 | C15H14N2OS | 66.64 | 5.22 | 10.36 | 11.86 | |
| 44 | 270.34 | 66.76 | 5.12 | 10.20 | 11.60 |
| 1c | H | 4-OCH3 | 75-80 | C15H14N2O2S | 62.92 | 4.93 | 9.78 | 11.20 | |
| 60 | 286.34 | 62.83 | 5.02 | 9.82 | 11.31 |
| 1d | H | 4-Cl | 110-112 | C14H11ClN2OS | 57.83 | 3.81 | 9.63 | 11.03 | 12.19 |
| 48 | 290.76 | 57.52 | 3.75 | 9.45 | 11.29 | 12.03 |
| 1f | H | 3-NO2 | 170-180 | C14H11N3O3S | 55.81 | 3.68 | 13.95 | 10.64 | |
| 34 | 301.31 | 55.65 | 3.54 | 13.97 | 10.75 |
| 1g | CH3 | H | 144-149 | C15H14N2OS | 66.64 | 5.22 | 10.36 | 11.86 | |
| 41 | 270.34 | 66.58 | 5.35 | 10.30 | 11.96 |
| 1h | CH3 | 4-CH3 | 163-165 | C16H16N2OS | 67.58 | 5.67 | 9.85 | 11.27 | |
| 34 | 284.37 | 67.41 | 5.80 | 9.71 | 11.09 |
| 1i | CH3 | 4-OCH3 | 150-153 | C16H16N2O2S | 63.98 | 5.37 | 9.33 | 10.67 | |
| 50 | 300.37 | 64.12 | 5.22 | 9.45 | 10.76 |
| 1j | CH3 | 4-Cl | 155-162 | C15H13ClN2OS | 59.11 | 4.30 | 9.19 | 10.52 | 11.63 |
| 40 | 304.79 | 59.03 | 4.12 | 9.26 | 10.64 | 11.47 |
| 1k | CH3 | 3-Br | 175-180 | C15H13BrN2OS | 51.59 | 3.75 | 8.02 | 9.18 | 22.88 |
| 33 | 349.24 | 51.74 | 3.73 | 7.95 | 9.30 | 22.65 |
| 1l | CH3 | 4-NO2 | 139-140 | C15H13N3O3S | 57.13 | 4.16 | 13.33 | 10.17 | |
| 35 | 315.34 | 57.33 | 4.31 | 13.21 | 10.00 |
| 1m | CH3 | 3-NO2 | 180-184 | C15H13N3O3S | 57.13 | 4.16 | 13.33 | 10.17 | |
| 32 | 315.34 | 57.29 | 4.27 | 13.17 | 10.05 |
Table 2.
– 1H-NMR chemical shifts of the 2-(4-subst.benzoylamino)thiobenzamidesa
Table 2.
– 1H-NMR chemical shifts of the 2-(4-subst.benzoylamino)thiobenzamidesa
| Compd. | H-2 | H-3 | H-4 | H-5 | H-o | H-m | NH2 | R1 | R2 |
|---|
| 1a | 8.25 | 7.27 | 7.54 | 7.48 | 8.00 | 7.61 | 9.93 + 10.37 | 11.23 | 7.67 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | bs;1H | m;1H |
| 1b | 8.26 | 7.25 | 7.52 | 7.47 | 7.88 | 7.42 | 9.92 + 10.33 | 11.15 | 2.44 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | bs;1H | s; 3H |
| 1c | 8.27 | 7.24 | 7.52 | 7.47 | 7.96 | 7.15 | 9.91 + 10.34 | 11.13 | 3.89 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | bs;1H | s; 3H |
| 1d | 8.17 | 7.28 | ←7.48 - 7.55→ | 7.99 | 7.69 | 9.89 + 10.32 | 11.20 | |
| m; 1H | m; 1H | m; 2H | m; 2H | m; 2H | 2×bs; 2H | bs;1H | |
| 1g | ← 7.18 - 7.30 → | 6.94 | 7.58 | 7.18-7.30 | 9.51 + 10.09 | 3.33 | 7.18- |
| m; 3H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | s; 3H | 7.30 |
| 1h | ← 7.18 - 7.28 → | 6.93 | 7.00 | 7.47 | 9.49 + 10.08 | 3.31 | 2.26 |
| m; 3H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | s; 3H | s; 3H |
| 1i | ← 7.18 - 7.30 → | 6.96 | 7.53 | 6.76 | 9.48 + 10.04 | 3.51 | 3.74 |
| m; 3H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | s; 3H | s; 3H |
| 1j | ← 7.23 - 7.30 → | 7.04 | 7.57 | 7.23-7.30 | 9.52 + 10.09 | 3.34 | |
| m; 3H | m; 1H | m; 2H | m; 2H | 2×bs; 2H | s; 3H | |
| 1l | ← 7.21 - 7.32 → | 8.06 | 7.76 | 9.36 + 10.01 | 3.38 | |
| m; 4H | m; 2H | m; 2H | 2×bs; 2H | s; 3H | |
Table 3.
– 1H-NMR chemical shifts of the 2-(3-subst.benzoylamino)thiobenzamides
Table 3.
– 1H-NMR chemical shifts of the 2-(3-subst.benzoylamino)thiobenzamides
| Compd. | H-2 | H-3 | H-4 | H-5 | H-8 | H-10 | H-11 | H-12 | NH2 | R1 |
|---|
| 1f | 8.10 | 7.31 | 7.52 - 7.57 | 8.81 | 8.49 | 7.91 | 8.40 | 9.86 + 10.28 | 11.33 |
| m; 1H | m; 1H | m; 2H | s; 1H | m; 1H | m; 1H | m; 1H | 2×bs; 2H | bs;1H |
| 1k | ←7.31 → | 7.24 | 7.05 | 7.83 | 7.58 | 7.18 | 7.49 | 9.58 + 10.15 | 3.36 |
| m; 2H | m; 1H | m; 1H | s; 1H | m; 1H | m; 1H | m; 1H | 2×bs; 2H | s; 3H |
| 1m | ←7.24 - 7.33 → | 8.34 | 8.18 | 7.54 | 7.93 | 9.39 + 10.04 | 3.40 |
| m; 4H | s; 1H | m; 1H | m; 1H | m; 1H | 2×bs; 2H | s; 3H |
Table 4.
– 13C-NMR chemical shifts of the 2-(4-subst.benzoylamino)thiobenzamides
Table 4.
– 13C-NMR chemical shifts of the 2-(4-subst.benzoylamino)thiobenzamides
| No. | C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C=S | C=O | C-i | C-o | C-m | C-p | R1 | R2 |
|---|
| 1a | 134.5 | 127.2 | 124.0 | 130.5 | 122.9 | 135.4 | 199.4 | 164.7 | 132.2 | 127.4 | 129.0 | 132.2 | | |
| 1b | 131.9 | 127.1 | 123.8 | 130.5 | 122.6 | 135.6 | 199.4 | 164.5 | 131.7 | 127.4 | 129.5 | 142.3 | | 21.2 |
| 1c | 131.8 | 127.0 | 123.6 | 131.0 | 122.5 | 135.6 | 199.3 | 164.7 | 126.5 | 129.2 | 114.2 | 162.3 | | 55.6 |
| 1d | 131.2 | 127.2 | 124.2 | 130.5 | 123.1 | 135.2 | 199.3 | 163.7 | 132.5 | 129.0 | 129.3 | 137.0 | | |
| 1g | 138.8 | 129.4 | 127.3 | 129.6 | 127.1 | 140.9 | 200.7 | 169.1 | 136.0 | 127.5 | 128.6 | 129.9 | 38.0 | |
| 1h | 139.2 | 129.4 | 127.3 | 129.8 | 127.0 | 141.1 | 200.7 | 169.1 | 133.1 | 128.0 | 128.8 | 138.9 | 38.0 | 21.0 |
| 1i | 138.9 | 129.6 | 127.3 | 130.8 | 127.0 | 141.4 | 200.7 | 168.9 | 128.1 | 129.6 | 112.8 | 160.2 | 38.3 | 55.2 |
| 1j | 139.0 | 129.7 | 127.6 | 129.8 | 127.3 | 140.7 | 200.4 | 168.1 | 134.3 | 130.5 | 130.5 | 134.9 | 38.1 | |
| 1l | 139.3 | 129.8 | 127.8 | 130.0 | 127.3 | 140.2 | 200.0 | 167.3 | 142.2 | 129.7 | 122.8 | 147.7 | 38.2 | |
Table 5.
– 13C-NMR chemical shifts of the 2-(3-subst.benzoylamino)thiobenzamides
Table 5.
– 13C-NMR chemical shifts of the 2-(3-subst.benzoylamino)thiobenzamides
| No. | C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 | C-8 | C-9 | C-10 | C-11 | C-12 | C=S | C=O |
|---|
| 1f | 133.7 | 127.4 | 124.7 | 130.6 | 122.3 | 136.0 | 134.7 | 123.7 | 148.0 | 126.5 | 130.4 | 133.7 | 199.4 | 162.8 |
| 1k | 138.9 | 129.7 | 127.5 | 129.9 | 127.4 | 140.5 | 138.2 | 129.6 | 120.9 | 132.4 | 131.4 | 127.4 | 200.5 | 167.5 |
| 1m | 139.3 | 129.8 | 127.7 | 130.0 | 127.4 | 140.3 | 137.4 | 123.5 | 147.0 | 124.4 | 129.2 | 134.9 | 200.1 | 166.8 |
Table 6.
– Melting points and elemental analyses of the 2-(3- and 4-subst.phenyl)quinazoline-4-thiones 2a-m
Table 6.
– Melting points and elemental analyses of the 2-(3- and 4-subst.phenyl)quinazoline-4-thiones 2a-m
| Compound | R1 | R2 | m.p. (°C) Yield (%) | Formula M.W. | Elemental analysis Calculated./Found (%) |
|---|
| C | H | N | S | X |
|---|
| 2a | H | H | 221-223a | C14H10N2S | 70.56 | 4.23 | 11.76 | 13.45 | |
| 88 | 238.30 | 70.39 | 4.39 | 11.82 | 13.49 |
| 2b | H | 4-CH3 | 221-222a | C15H12N2S | 71.40 | 4.79 | 11.10 | 12.71 | |
| 86 | 252.33 | 71.15 | 4.72 | 11.21 | 12.57 |
| 2c | H | 4-OCH3 | 200-201a | C15H12N2OS | 67.14 | 4.51 | 10.44 | 11.95 | |
| 85 | 268.33 | 67.20 | 4.31 | 10.35 | 11.65 |
| 2d | H | 4-Cl | 287-289a | C14H9ClN2S | 61.65 | 3.33 | 10.27 | 11.75 | 13.00 |
| 83 | 272.75 | 61.52 | 3.35 | 10.11 | 11.68 | 13.03 |
| 2e | H | 4-NO2 | 250-252b | C14H9N3O2S | 59.35 | 3.20 | 14.83 | 11.32 | |
| 89 | 283.30 | 59.10 | 3.30 | 15.04 | 11.39 |
| 2f | H | 3-NO2 | 263-265 | C14H9N3O2S | 59.35 | 3.20 | 14.83 | 11.32 | |
| 84 | 283.30 | 59.27 | 3.24 | 14.93 | 11.20 |
| 2g | CH3 | H | 197-199 | C15H12N2S | 71.40 | 4.79 | 11.10 | 12.71 | |
| 91 | 252.33 | 71.20 | 4.70 | 11.21 | 12.86 |
| 2h | CH3 | 4-CH3 | 237-239 | C16H14N2S | 72.15 | 5.30 | 10.52 | 12.04 | |
| 87 | 266.36 | 72.15 | 5.39 | 10.38 | 12.41 |
| 2i | CH3 | 4-OCH3 | 95-98 | C16H14N2OS | 68.06 | 5.00 | 9.92 | 11.35 | |
| 84 | 282.35 | 67.95 | 5.10 | 9.86 | 11.48 |
| 2j | CH3 | 4-Cl | 249-251 | C15H11ClN2S | 62.82 | 3.87 | 9.77 | 11.18 | 12.36 |
| 89 | 286.77 | 62.74 | 3.93 | 9.73 | 10.95 | 12.29 |
| 2k | CH3 | 3-Br | 252-254 | C15H11BrN2S | 54.39 | 3.35 | 8.46 | 9.68 | 24.12 |
| 81 | 331.22 | 54.77 | 3.41 | 8.38 | 9.46 | 24.00 |
| 2l | CH3 | 4-NO2 | 264-266 | C15H11N3O2S | 60.59 | 3.73 | 14.13 | 10.78 | |
| 85 | 297.33 | 60.05 | 3.87 | 14.42 | 10.92 |
| 2m | CH3 | 3-NO2 | 278-280 | C15H11N3O2S | 60.59 | 3.73 | 14.13 | 10.78 | |
| 80 | 297.33 | 60.87 | 3.44 | 13.96 | 10.86 |
Table 7.
– 1H-NMR chemical shifts of the 2-(4-subst.phenyl)quinazoline-4-thiones
Table 7.
– 1H-NMR chemical shifts of the 2-(4-subst.phenyl)quinazoline-4-thiones
| Compd. | H-2 | H-3 | H-4 | H-5 | H-o | H-m | R1 | R2 |
|---|
| 2aa | 8.66 | 7.58-7.67 | 7.94 | 7.82 | 8.20 | 7.58-7.67 | 13.96 | 7.58-7.67 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | bs; 1H | m; 1H |
| 2b | 8.65 | 7.63 | 7.94 | 7.81 | 8.14 | 7.42 | 13.88 | 2.46 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | bs; 1H | s; 3H |
| 2c | 8.64 | 7.60 | 7.92 | 7.78 | 8.22 | 7.15 | 13.80 | 3.90 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | bs; 1H | s; 3H |
| 2d | 8.66 | 7.63-7.70 | 7.94 | 7.83 | 8.24 | 7.63-7.70 | 14.02 | |
| m, 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | bs; 1H | |
| 2e | 8.68 | 7.69 | 7.99 | 7.87 | ← 8.44 → | 14.25 | |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 4H | bs; 1H | |
| 2g | 8.67 | 7.80-7.82 | 7.99 | 7.88 | ←– 7.63 - 7.68 –→ | 3.75 | 7.80-7.82 |
| m; 1H | m; 2H | m; 1H | m; 1H | m; 4H | s; 3H | m; 2H |
| 2h | 8.66 | 7.66 | 7.96 | 7.87 | 7.72 | 7.45 | 3.79 | 2.47 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | s; 3H | s; 3H |
| 2I | 8.66 | 7.65 | 7.97 | 7.87 | 7.82 | 7.19 | 3.83 | 3.92 |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | s; 3H | s; 3H |
| 2j | 8.67 | 7.67 | 7.99 | 7.89 | 7.86 | 7.72 | 3.77 | |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | s; 3H | |
| 2l | 8.69 | 7.70 | 8.02 | 7.93 | 8.48 | 8.10 | 3.74 | |
| m; 1H | m; 1H | m; 1H | m; 1H | m; 2H | m; 2H | s; 3H | |
Table 8.
– 1H-NMR chemical shifts of the 2-(3-subst.phenyl)quinazoline-4-thiones
Table 8.
– 1H-NMR chemical shifts of the 2-(3-subst.phenyl)quinazoline-4-thiones
| Compd. | H-2 | H-3 | H-4 | H-5 | H-9 | H-11 | H-12 | H-13 | R1 |
|---|
| 2f | 8.65 | 7.67 | 7.96 | 7.88 | 9.04 | 8.65 | 7.88 | 8.48 | 14.27 |
| m; 2H | m; 1H | m; 1H | m; 2H | m; 1H | m; 2H | m; 2H | m; 1H | bs; 1H |
| 2k | 8.67 | 7.68 | 8.00 | 7.88 | 8.00 | 7.82 | 7.60 | 7.88 | 3.77 |
| m; 1H | m; 1H | m; 2H | m; 2H | m; 2H | m; 1H | m; 1H | m; 2H | s; 3H |
| 2m | 8.69 | 7.70 | 8.02 | 7.94 | 8.69 | 8.51 | 7.94 | 8.27 | 3.78 |
| m; 2H | m; 1H | m; 1H | m; 2H | m; 2H | m; 1H | m; 2H | m; 1H | s; 3H |
Table 9.
– 13C-NMR chemical shifts of the 2-(4-subst.phenyl)quinazoline-4-thiones
Table 9.
– 13C-NMR chemical shifts of the 2-(4-subst.phenyl)quinazoline-4-thiones
| No. | C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C=S | C-7 | C-i | C-o | C-m | C-p | R1 | R2 |
|---|
| 2aa | 127.8 | 129.5 | 128.3 | 135.7 | 128.2 | 144.5 | 188.0 | 151.7 | 132.3 | 128.7 | 128.6 | 131.7 | | |
| 2b | 127.6 | 129.4 | 128.2 | 135.5 | 127.9 | 144.5 | 188.0 | 151.5 | 129.4 | 128.5 | 129.2 | 141.8 | | 21.1 |
| 2c | 127.4 | 129.4 | 128.0 | 135.5 | 127.6 | 144.5 | 187.9 | 151.1 | 124.2 | 130.3 | 114.0 | 162.1 | | 55.6 |
| 2d | 127.7 | 129.4 | 128.2 | 135.5 | 128.1 | 144.3 | 188.2 | 150.8 | 131.3 | 130.4 | 128.6 | 136.4 | | |
| 2e | 128.0 | 129.4 | 128.7 | 135.6 | 128.5 | 144.1 | 188.1 | 150.3 | 138.4 | 130.2 | 123.6 | 149.1 | | |
| 2g | 128.1 | 130.6 | 127.5 | 134.7 | 117.7 | 137.5 | 199.0 | 154.9 | 134.5 | 129.4 | 128.6 | 130.8 | 38.8 | |
| 2h | 128.1 | 130.6 | 127.3 | 134.6 | 117.6 | 137.6 | 198.9 | 154.9 | 131.5 | 129.1 | 129.6 | 140.9 | 38.9 | 21.2 |
| 2i | 128.0 | 130.5 | 127.1 | 134.4 | 117.6 | 137.7 | 198.7 | 154.5 | 126.2 | 131.8 | 113.9 | 161.4 | 39.2 | 55.6 |
| 2j | 128.2 | 130.5 | 127.5 | 134.6 | 117.6 | 137.4 | 198.9 | 153.9 | 133.3 | 131.4 | 128.6 | 135.6 | 38.7 | |
| 2l | 128.3 | 130.6 | 127.8 | 134.9 | 117.7 | 137.3 | 199.0 | 153.2 | 140.6 | 130.9 | 123.7 | 146.6 | 38.6 | |
Table 10.
– 13C-NMR chemical shifts of the 2-(3-subst.phenyl)quinazoline-4-thiones
Table 10.
– 13C-NMR chemical shifts of the 2-(3-subst.phenyl)quinazoline-4-thiones
| No. | C-1 | C-2 | C-3 | C-4 | C-5 | C-6 | C-7 | C-8 | C-9 | C-10 | C-11 | C-12 | C-13 | C=S |
|---|
| 2f | 128.0 | 129.4 | 128.5 | 135.4 | 128.4 | 144.1 | 150.0 | 134.0 | 123.5 | 147.8 | 126.0 | 130.2 | 134.9 | 188.1 |
| 2k | 128.3 | 130.6 | 127.7 | 134.8 | 117.7 | 137.4 | 153.5 | 136.7 | 131.9 | 121.7 | 133.6 | 130.8 | 128.4 | 199.0 |
| 2m | 128.1 | 130.6 | 127.7 | 134.8 | 117.8 | 137.5 | 153.0 | 136.1 | 124.5 | 147.7 | 125.3 | 130.3 | 135.7 | 199.0 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}








