Synthesis of 2,4,6-Tris(4-N-isopropylamidinophenyl)pyrimidine trihydrochloride

1
Department of Organic Chemistry, Faculty of Chemical Engineering and Technology, University of Zagreb, 10000 Zagreb, Croatia
2
Department of Chemistry and Center for Biotechnology and Drug Design, Georgia State University, Atlanta, Georgia 30303-3083, USA
*
Authors to whom correspondence should be addressed.
Molecules 2001, 6(5), 477-480; https://doi.org/10.3390/60500477
Submission received: 20 January 2001
/
Accepted: 17 March 2001
/
Published: 30 April 2001
{kind=link}
Abstract
:The synthesis of 2,4,6-tris(4-N-isopropylamidinophenyl)pyrimidine from 1,3-di(4-bromophenyl)propen-3-one and 4-bromobenzamidine is reported.
Introduction
In previous reports, we have described the synthesis, DNA binding results and significant anti-pneumocystic activity for a series of dicationic diarylpyrimidines [1,2,3]. We demonstrated that by careful manipulation of structure highly DNA selective molecules could be developed. This report describes the synthesis of a new tricationic triarylpyrimidine which is expected to show strong DNA binding affinity.
Results and Discussion
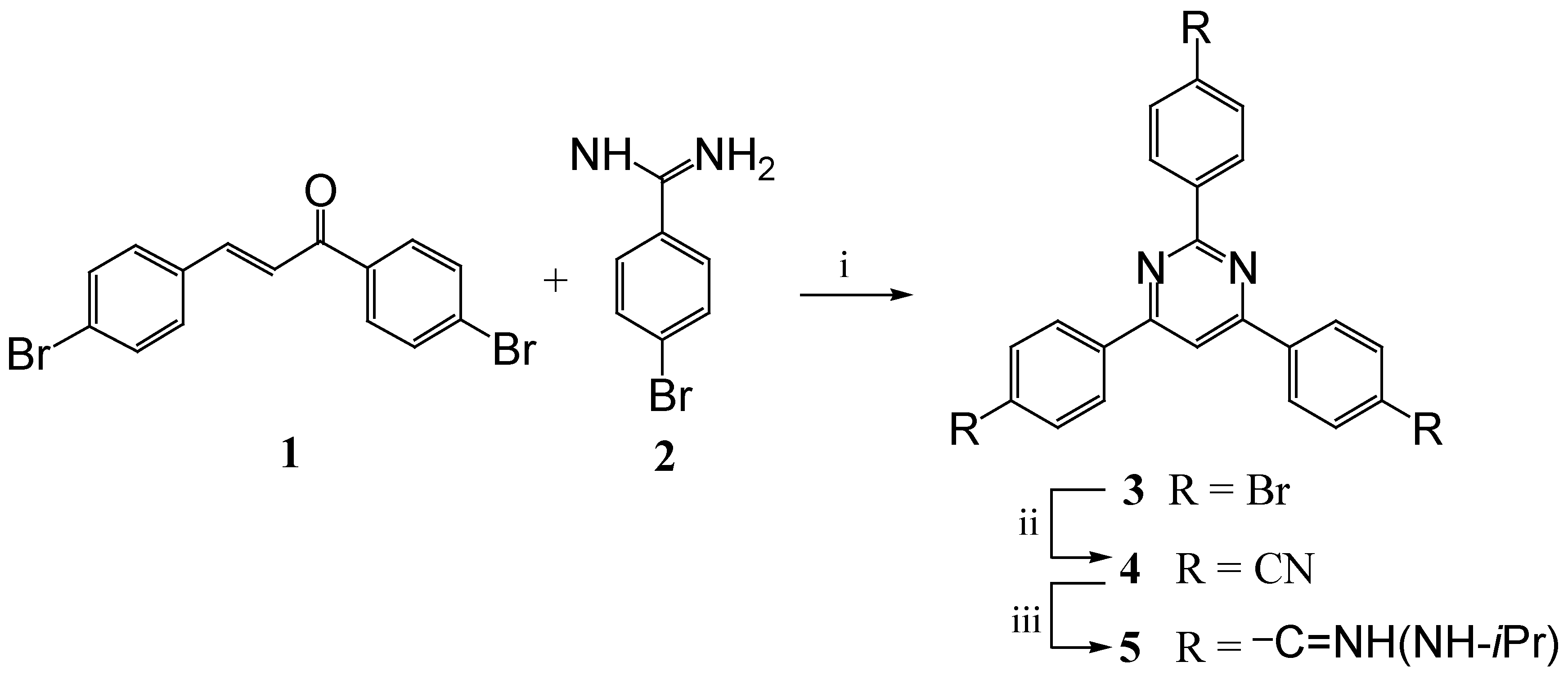
The synthetic route employed to make 2,4,6-tris(4-N-isopropylamidinophenyl)pyrimidine trihydro-chloride (5) is outlined in Scheme 1. The key intermediate 2,4,6-tris(4-bromophenyl)pyrimidine (3) was obtained in good yield by base-promoted condensation between 1,3-di(4-bromophenyl)propen-3-one (1) and 4-bromobenzamidine benzensulfonate (2), using an approach similar to the one described by Dodson and Seyler [4]. This reaction sequence uses two moles of the α,β-unsaturated ketone, one mole becomes incorporated in the pyrimidine ring and the other is employed as the oxidizing agent for the intermediate dihydropyrimidine.
Tribromo compound 3 was converted into the corresponding tris-nitrile 4, which was then transformed into the tricationic target compound employing the standard approach [1,2].
Scheme 1.

i) KOH, EtOH; ii) CuCN, quinoline; iii) HCl, EtOH; H2N-iPr, EtOH.
Experimental
General
Melting points were recorded using a Thomas Hoover (Uni-Melt) capillary melting point apparatus and are uncorrected. 1H-NMR and 13C-NMR spectra were recorded employing a Varian GX400 spectrometer and chemical shifts (δ) are in ppm relative to TMS. High resolution mass spectra were recorded with a VG Instruments 70-SE spectrometer (Georgia Institute of Technology, Atlanta, GA); others were recorded by a Shimadzu GC-MS 5000 instrument at 70 eV chamber voltage on a direct inlet system. Elemental analysis were obtained from Atlantis Microlab Inc. (Norcross, GA). All chemicals and solvents were purchased from Aldrich Chemical Co. or Fisher Scientific.
2,4,6-Tris(4-bromophenyl)pyrimidine (3)
1,3-Di(4-bromophenyl)propen-3-one (1), prepared by a standard literature method [5] (15 g, 41 mmol) and 4-bromobenzamidine benzensulfonate [1] (7.32 g, 20.5 mmol) was dissolved in ethanol (600 mL) and a solution of potassium hydroxide (2.3 g, 41 mmol) in ethanol (200 mL) was added. The reaction mixture was refluxed for 3 hr, cooled and precipitate was collected by filtration and washed with water. White solid was recrystallized from glacial acetic acid to yield 9.78 g (87.6%) of white crystals; Mp: >290 °C; MS (EI, 70eV): 542/544/546/548 (peaks in the molecular ion region); 1H-NMR (400 MHz, CDCl3): 8.55 (d, 2H, J = 8.5 Hz); 8.14 (d, 4H, J = 8.8 Hz); 7.95 (s, 1H); 7.70 (d, 4H, J = 8.6 Hz); 7.66 (d, 2H, J = 8.8 Hz); 13C-NMR (100 MHz, CDCl3): 164.2; 164.1; 136.9; 136.3; 132.3; 131.8; 130.2; 128.8; 125.8; 109.9; Anal. calcd. for C22H13Br3N2 (545.07): C 48.47, H 2.40, N 5.14. Found: C 48.36, H 2.42, N 5.08.
2,4,6-Tris(4-cyanophenyl)pyrimidine (4)
A mixture of 2,4,6-tris(4-bromophenyl)pyrimidine (3) (5 g, 9.17 mmol) and copper(I)cyanide (4.1 g, 45.9 mmol) in freshly distilled quinoline (75 mL) was refluxed for 3 hr [1,2]. After cooling the reaction mixture was poured into ether (100 mL), filtered and solid was washed with ether and water, and than extracted in a Soxhlet apparatus using acetone as a solvent. Acetone solution was passed through a short alumina column to remove copper salts. Solvent was evaporated and solid was crystallized from acetone to yield 1.95 g (55.6%) of white fluffy needles; Mp: >290 °C; MS (EI, 70 eV) : 383 (M+); 1H-NMR (400 MHz, DMSO-d6): 8.88 (s, 1H); 8.83 (d, 2H, J = 8.3 Hz); 8.73 (d, 4H, J = 8.5 Hz); 8.13 (d, 4H, J = 8.3 Hz); 8.09 (d, 2H, J = 8.3 Hz); 13C-NMR (100 MHz, DMSO-d6): 163.1; 162.0; 140.8; 140.0; 132.5; 132.4; 128.5; 128.1; 118.2; 118.0; 113.5; 113.3; 112.9; Anal. calcd. for: C25H13N5 (383.39): C 78.31, H 3.42, N 18.27. Found: C 78.27, H 3.42, N 18.19.
2,4,6-Tris(4-N-isopropylamidinophenyl)pyrimidine trihydrochloride (5)
A stirred suspension of 4 (0.36 g, 0.94 mmol) in anhydrous dioxane (50 mL) and absolute ethanol (20 mL) was chilled to 0-5 °C, and was saturated with HCl(g). The contents were stirred at room temperature until IR spectra indicated the disappearance of the nitrile peak. The crude triimidate ester was collected by filtration and washed with dry ether. To a stirred suspension of crude triimidate ester in absolute ethanol (100 mL) freshly distilled isopropylamine (0.95 g, 16 mmol) was added and the mixture was stirred for 72 hr at room temperature. Solvent was partially evaporated and acetone was added to the residue. Solid was collected by filtration, washed with acetone and dry ether. The free base was suspended in absolute ethanol saturated with HCl (50 mL) and heated at reflux for 4 hr. After cooling, the solid was collected by filtration, washed with acetone and dried in vacuum at 90 °C for 48 hr to yield 0.23 g (35%) of white powder; Mp: > 290 °C; MS (FAB): 561.3 (M++1); HRMS (FAB): calcd. for C34H40N8: 561.3454 (M++1). Found: 561.3451; 1H-NMR (400 MHz, DMSO-d6): 9.85 (bs, 3H, NH); 9.68 (bs, 3H, NH); 9.29 (bs, 3H, NH); 8.91 (s, 1H, H-5); 8.84 (d, 2H, J = 8.5 Hz, H-a'); 8.77 (d, 4H, J = 8.4 Hz, H-a); 8.02 (d, 4H, J = 8.6 Hz, H-b); 7.99 (d, 2H, J = 8.9 Hz, H-b'); 4.15 (m, 3H, J = 6.6 Hz, CH); 1.34 (d, 18H, J = 6.3 Hz, CH3); 13C-NMR (100 MHz, DMSO-d6+D2O): 163.8; 162.9; 161.6; 161.3; 141.2; 140.2; 132.3; 132.0; 129.0; 128.9; 128.4; 128.0; 112.7; 45.3; 21.4; Anal. calcd. for C34H40N8·3HCl·1.5H2O (697.14): C 58.57, H 6.65, N 16.07. Found: C 58.32, H 6.63, N 15.89.
References
- Kumar, A.; Boykin, D. W.; Wilson, W. D.; Jones, S. K.; Bender, B. C.; Dykstra, C. C.; Hall, J. E.; Tidwell, R. R. Eur. J. Med. Chem 1996, 31, 767–773. [PubMed]
- Boykin, D. W.; Kumar, A.; Bajić, M.; Xiao, G.; Wilson, W. D.; Bender, B. C.; McCurdy, D. R.; Hall, J. E.; Tidwell, R. R. Eur. J. Med. Chem 1997, 32, 965–972.
- Kumar, A.; Zhao, M.; Wilson, W. D.; Boykin, D. W. Bioorg. Med. Chem. Lett. 1994, 4, 2913–2918.
- Dodson, R. M.; Seyler, J. K. J. Org. Chem 1951, 16, 461–465.
- Kohler, E. P.; Chadwell, H. M. Org. Synth. Coll. Vol. 1 1941, 78–80.
- Sample availability: Not available.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes
Share and Cite
MDPI and ACS Style
Bajić, M.; Boykin, D.W. Synthesis of 2,4,6-Tris(4-N-isopropylamidinophenyl)pyrimidine trihydrochloride. Molecules 2001, 6, 477-480. https://doi.org/10.3390/60500477
AMA Style
Bajić M, Boykin DW. Synthesis of 2,4,6-Tris(4-N-isopropylamidinophenyl)pyrimidine trihydrochloride. Molecules. 2001; 6(5):477-480. https://doi.org/10.3390/60500477
Chicago/Turabian StyleBajić, Miroslav, and David W. Boykin. 2001. "Synthesis of 2,4,6-Tris(4-N-isopropylamidinophenyl)pyrimidine trihydrochloride" Molecules 6, no. 5: 477-480. https://doi.org/10.3390/60500477