Reactions of 5-Aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines with Nitrile Oxides

Chemistry Department, Faculty of Science, Ain Shams University, Abbassia, Cairo, Egypt
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(6), 510-518; https://doi.org/10.3390/60600510
Submission received: 4 July 2000
/
Revised: 13 January 2001
/
Accepted: 30 April 2001
/
Published: 31 May 2001
Abstract
:E,Z-5-Aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines (3a-c) react with 4-methoxy and 4-chlorophenylnitrile oxides (4a and b) in pyridine solution to afford one or more of the following compounds: Z-3, Z-2,4-dioxo analogues 5 and 3,6-diaryl- 1,4,2,5-dioxadiazines (6a-b). The interconversion route is discussed and the structures of all of the synthesised compounds are proven by microanalytical and spectral data.
Introduction
Several reports on the chemistry of 4-thiazolidinones have been published [1,2,3,4,5,6], but literature reports on reactions of 4-oxo-2-thioxo-thiazolidines with dipolar species are rather limited and treat the problem of alkylation of the 3-unsubstituted derivatives with diazomethane [7,8] and cycloaddition of nitrilimines to the thiono function of 5-aroyl-methylene-3-phenyl-4-oxo-2-thioxo-1,3-thiazolidine [9]. The intention of the present work was to study the reactivity of thiono as well as exocylic double bond functions in E,Z-5-aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines (3a-c) towards 4-methoxyphenyl- and 4-chlorophenylnitrile oxides (4a,b). Thus a series of E,Z-5-aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3- thiazolidines (3a-c) has been synthesised and subsequently reacted with 4-methoxyphenyl- and 4- chlorophenylnitrile oxides (4a,b).
Results and Discussion
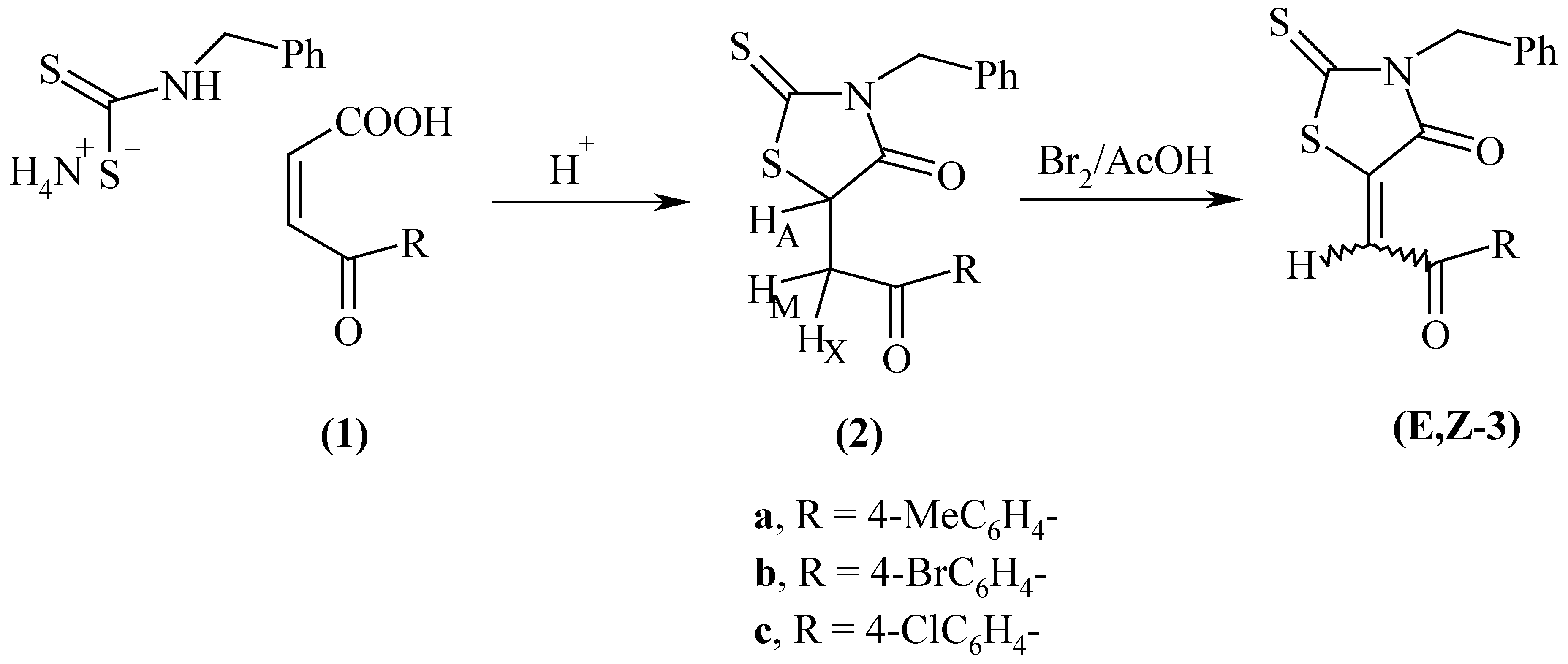
Compounds E,Z-3a-c were synthesised by a method similar to that of Nagase [10] by treating 5-(2- aryl-2-oxoethyl)-4-oxo-2-thioxo-1,3-thiazolidines (2a-c) (obtained in turn by reaction of 3-aroylacrylic acids (1a-c) with ammonium benzyldithiocarbamate), with bromine as shown in Scheme 1. Omar et al. [2] studied the stereochemistry of compounds 3 and reported the formation of E- and Z- stereoisomers with the predominance of the former one.
Scheme 1.

Our intent was to perform the reactions in pyridine as, on the one hand, it is able to dissolve the starting thiazolidinones which are sparingly soluble in most organic solvents, and, on the other hand, it can be used to liberate the required nitrile oxide in situ from stable α-hydroximinobenzyl chlorides precursors.
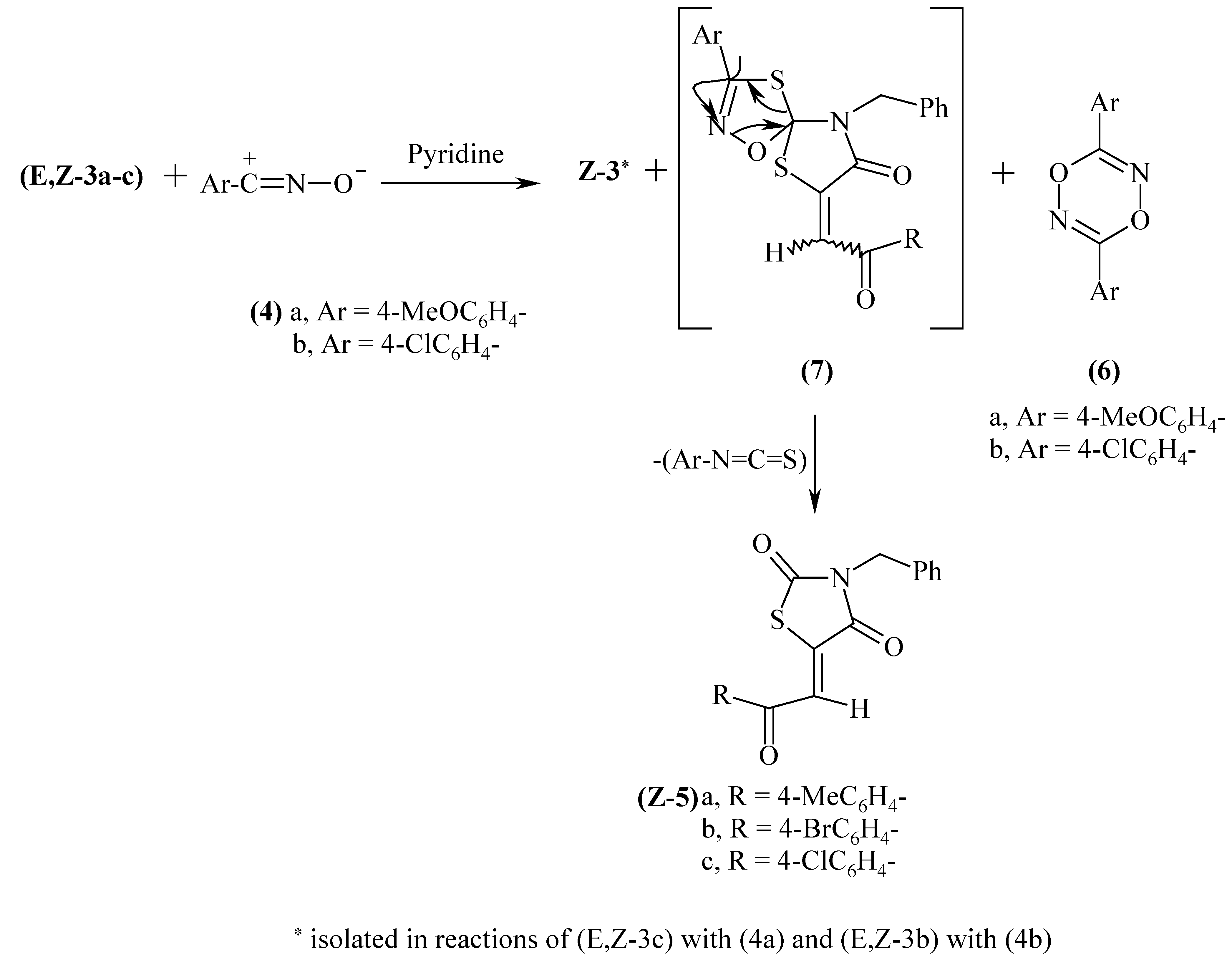
Nitrile oxides 4a and 4b, produced in situ upon addition of α-hydroximino-4-methoxy- and α- hydroximino-4-chlorobenzyl chlorides to pyridine reacted with E,Z-3a-c to afford the Z-isomers of either of the starting 4-oxo-2-thioxo- compounds Z-3 or the Z-2,4-dioxo- analogues 5, or a mixture of them, along with the corresponding head to tail dimerised product, namely 3,6-di-(4-methoxyphenyl)- 1,4,2,5-dioxadiazine (6a), in the former case and the 4-chlorophenyl counterpart (6b), in the latter case, as outlined in Scheme 2. Small amounts of the unreacted starting materials somewhat enriched with Z- isomers were isolated in all the studied examples. (cf. Table 1).
The structures of the isomerized 4-oxo-2-thioxo-1,3-thiazolidines (Z-3b and c) and the respective 2,4-dioxo counterparts (Z-5a-c) were deduced from microanalytical, IR, 1H-NMR and MS spectral data (cf. Table 2 and Table 3). All the IR spectra of compounds 5 show the complex carbonyl pattern extending to 1750 cm-1 consistent with the stretching vibrations of coupled carbonyl groups, whereas those of 3 show the carbonyl absorptions at mostly lower frequency values not exceeding 1715 cm-1. Their EI-mass spectra exhibit parent peaks at m/e 91, molecular ion peaks and M.+-CS(CO) [A] and M.+-PhCH2NCS(O) [B] fragments. The structures of 6a and 6b, which were believed to be the head to the tail dimerised products, was confirmed by comparison (m.p and IR) with authentic samples [11].
Scheme 2.

Configurational assignments of compounds 3 and 5 were based exclusively on 1H-NMR spectroscopy by comparing the observed chemical shift values of the olefinic protons with the incremented values [12]; the olefinic protons of the Z-isomers are relatively deshielded by the 4-oxo- groups compared with the E-counterparts. The Z-configuration was assigned to 3b and 3c by comparing with the starting E-counterparts and to 5a by comparing with a sample previously prepared [1] upon treating a solution of 2a in glacial acetic acid with bromine.
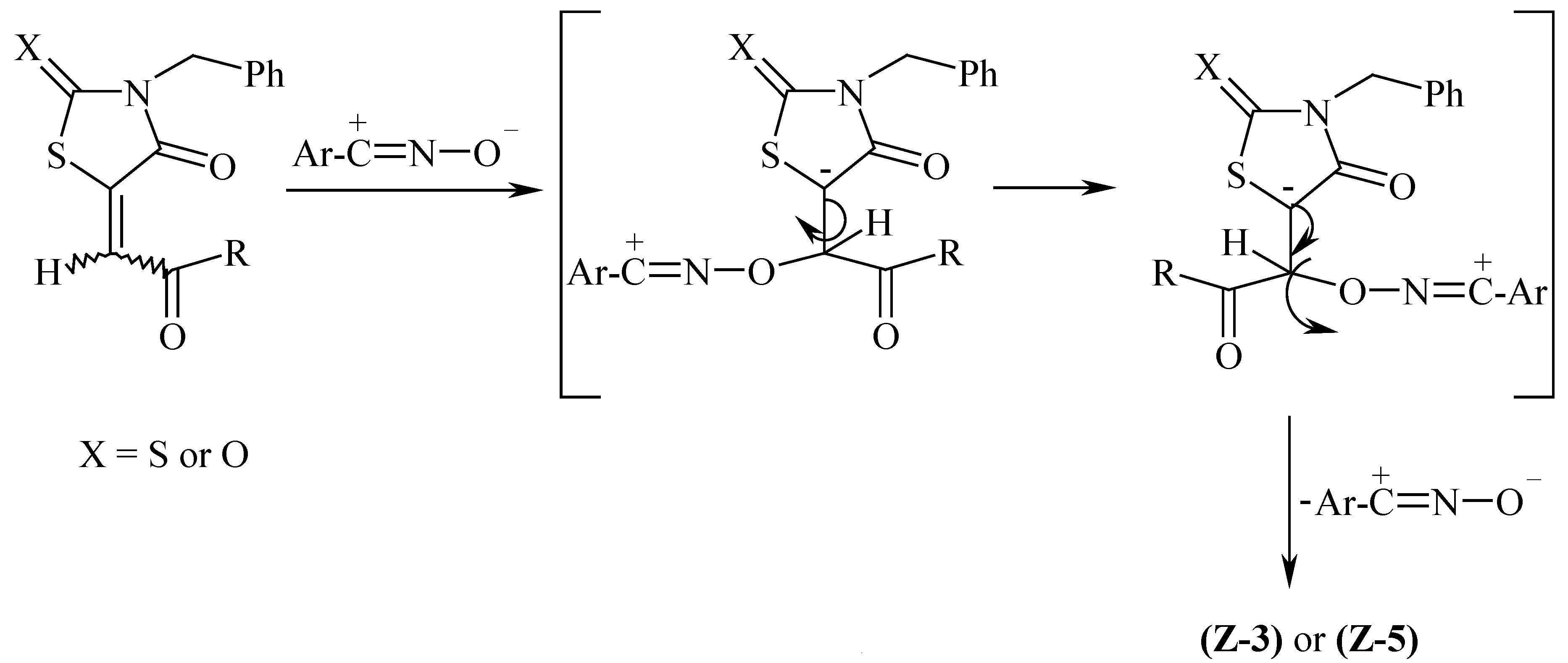
A role for pyridine alone in the isomerisation process can be ruled out as compounds E,Z-3a-c are recovered without detectable configurational change upon refluxing in this solvent. The larger proportions of Z-isomers observed in the compounds prepared as compared to the starting materials indicates that isomerisation has occured during the reactions. The great stability of the Z-isomers as compared with the E-counterparts is probably due to steric considerations Although isomerisation may be explained according to the hypothesis of formation of zwitterionic or biradical intermediates [13], it is better explained in terms of successive addition and elimination of nitrile oxide at the β-terminus of the exocyclic α,β-unsatured carbonyl system of compounds 3 as outlined in Scheme 3. The instability of the hypothetically formed adducts may be attributed to a sterically hindered transition state. The negative results reported for the reaction of tri- and tetra-alkyl ethylenes with nitrile oxides [14] seem to be in accordance with our results.
Scheme 3.

The formation of the 2,4-dioxo compounds 5 rather than the spiranes 7; which could be produced by the attack of nitrile oxides at the thiono group of compounds 3 could be rationalised in terms of decomposition of the 1,4,2-oxathiazole ring of the spirane ring system by expelling 4-chlorophenyl- or 4- methoxyphenylisothiocyanates. The decomposition has occured most likely via a radical reaction as it has been reported by Husigen et al [15] that all the 1,4,2-oxathiazoles which are formed via cycloaddition of nitrile oxides to thiocarbonyl compounds decompose exothermically at 90-150°C to form isothiocyanates and the oxygen analogues of the thiocarbonyl compounds. The molecular rearrangement leading to the isothiocyanate probably proceeds concurrently with the ring opening [15] (Scheme 2). The preference for the nitrile oxides to react with C=S rather than the C=O groups may be attributed to the polarizability of the sulfur function which is manifested by the great readiness with which several thiono containing compounds react with dipolar species [16]. Formation of stable spiranes is reported upon reacting 2-thioxo-1,3,4-thiadiazole with nitrile imines [16].
Experimental
General
All melting points are uncorrected. IR spectra were measured on a Unicam SP1200 spectrometer as KBr discs. 1H-NMR spectra were recorded in d6-DMSO on a Varian Gemini 200 MHz instrument with chemical shifts (δ) expressed in ppm downfield from TMS. Mass spectra were recorded on Shimadzu GC-MS-Qp 1000 EX instrument operating at 70 ev. Column chromatography and TLC were run on Silica Gel Woelm, activity 113/30 mm according to Brockmann & Schodder and Silica Gel 60 F254 (Merck) aluminium backed TLC sheets.
Preparation of Starting Materials 3-Benzyl-5-[2-(4-bromophenyl)-2-oxoethyl]-4-oxo-2-thioxo-1,3-thiazolidine (2b).
Ammonium benzyl-dithiocarbamate (2.2 g, 11 mmol) in ethanol (10 mL) was added dropwise to a stirred solution of 3-(4-bromobenzoyl) propenoic acid [17] (2.55 g, 10 mmol) in ethanol (10 mL) at room temperature. Concentrated hydrochloric acid (3 mL) was added portionwise to the stirred reaction mixture after 30 min. and the precipitated solid was filtered and recrystallised from benzene-light petroleum (b.p. 60-80°C) to give 2b [1,18].
E,Z-3-Benzyl-5-(4-bromobenzoylmethylene)-4-oxo-2-thioxo-1,3-thiazolidine (3b).
Bromine (20 mmol) was added to a solution of 2b (3.4 g, 10 mmol) in glacial acetic acid (20 mL) and the resulting mixture was warmed on a water bath for 5 min. After cooling the reaction mixture was poured into cold water and filtered. The crude orange product (3.34 g, 80%) was recrystallised from glacial acetic acid to give E,Z-3b.
The analogous compounds 3-benzyl-5-[2-(4-methylphenyl)-2-oxoethyl]-4-oxo-2-thioxo-1,3-thiazolidine (2a) and 3-benzyl-5-[2-(4-chlorophenyl)-2-oxoethyl]-4-oxo-2-thioxo-1,3-thiazolidine (2c) [18] and E,Z- 3-benzyl-5-(4-methylphenylmethylene)-4-oxo-2-thioxo-1,3-thiazolidine (3a) and E,Z-3-benzyl-5-(4- chlorophenylmethylene)-4-oxo-2-thioxo-1,3-thiazolidine (3c) [1,10] were prepared by the same methods mentioned above for 2b and E,Z-3b.
Reactions of Compounds E,Z-3a-c with Arylnitrile Oxides 4a and b: General Procedure
Powdered α-hydroximino-4-chlorobenzyl chloride (4a) [19] or the 4-methoxybenzyl counterpart (4b) [20] (25 mmol) was added to a suspension of each of E,Z-3a-c (10 mmol) in anhydrous pyridine (10 mL) and the whole mixture was refluxed for 10 hrs. during which it acquired a violet colouration. The solid which precipitated after allowing the reaction mixture to stand at room temperature overnight was filtered off, washed with small portion of ether and chromatographed or recrystallised from an appropriate solvent to give Z-3 and/or Z-5 along with starting materials slightly enriched with the Z-configured isomers E,Z-3.
The crude product (2.82 g, 80%) obtained upon treating E,Z-3a with 4b was crystallized from dioxane to give E,Z-3a as yellow crystals (0.71 g, 20%), m.p. 210-212°C containing ca. 20% of the Z- isomer. Dilution of the dioxane mother liquors gave Z-5a as brownish red crystals (1.77 g, 50%), m.p. 179-181°C (from dilute dioxane).
The reaction mixture of E,Z-3b with 4b was concentrated (final volume ca. 5 mL), chromatographed over silica gel while monitoring by TLC. Elution with light petroleum (b.p. 40-60°C)-ether mixture (4:1 v/v) gave Z-3b (2.3 g, 55%) as red needles (from benzene). Elution with light petroleum (b.p. 40-60°C)- ether mixture (1:1 v/v) gave Z-5b (0.4 g, 10%) as brownish red crystals (from benzene-methanol). Elution with pure ether gave E,Z-3b (0.63 g, 15%) containing 30% of the Z- isomer.
The crude product (2.98 g, 80%) which was obtained upon reacting E,Z-3c with 4b was crystallised from dioxane to give E,Z-3c (0.75 g, 20%) containing 90% of the Z-isomer as orange crystals m.p. 248- 250°C. On leaving the dioxane mother liquor to stand at room temperature for 24 hrs., it precipitated a yellow solid (1.96 g, 55%) which was recrystallised from benzene-methanol to give Z-5c as yellow crystals m.p. 226-228°C.
Recrystallisation of the crude product (1.49 g, 40%) obtained from the reaction of E,Z-3c with 4a from dioxane precipitated successively E,Z-3c (0.6 g, 16%) containing 10% of the Z-isomer and Z-3c (0.67 g, 18%).
Upon allowing the water diluted original pyridine mother liquors to stand at room temperature for 48 hrs. therefrom precipitated colourless products which were recrystallised from dioxane to give 45% of 3,6-di-(4-methoxyphenyl)-1,4,2,5-dioxadiazine (6a), m.p. 178-180°C, undepressed on admixture with an authentic sample [11] in all cases when 4a was used and 5-10% of 3,6-di-(4-chlorophenyl)-1,4,2,5- dioxadiazine (6b), m.p. 222-224°C undepressed on admixture with an authentic sample [11], when 4b was the reactant.
Action of pyridine on E,Z-3a-c
A suspension of each of E,Z-3a-c (3 mmol) in pyridine (10 mL) was refluxed for 15 hrs. while the solution acquired a violet colouration. The crude product which precipitated upon leaving the reaction mixture to stand at room temperature overnight (90-95%) was recrystallised from dioxane to give 90% of each E,Z-3a-c with similar E/Z ratios as the starting materials.

{kind=link}
{kind=link}
{kind=link}
| Reactants | Products (% yield)** | ||||
| (E,Z)*-3 | Nitrile oxide | (E,Z)*-3 [Z%]*** | (Z)*-3 | (Z)*-5 | 6 |
| 3c | 4a | 3c(16)[10] | 3c(18) | --- | 6a(45) |
| 3a | 4b | 3a(20)[20] | --- | 5a(50) | 6b(7) |
| 3b | 4b | 3b(15)[30] | 3b(55) | 5b(10) | 6b(5) |
| 3c | 4b | 3c(20)[90] | --- | 5c(55) | 6b(10) |
- * configurational assignment is based on 1H-NMR spectroscopy.
- ** yield of actually isolated compounds
- *** % of the Z-isomer in the E,Z-mixture.
| Compd. No. | MP (°C) | 1H-NMR Spectral data [δ-values ppm] | |||||
| Olefinic | AroylH* | N-CH2Ph | N-CH2Ph | Me | |||
| H | 2Ha | 2Hb | 5H | 2H | 3H | ||
| 2b a [1,18] | 128-129 | --- | 8.020(db) | 7.703(db) | 7.205(br.s) | 5.15 (br s) | -- |
| E,Z-3a[1] | 215-217 | 7.900(sc) | 7.980(dd) | 7.350-7.050 (m) | 5.300 (br sc) | 3.250 (s) | |
| 8.265(sc) | 4.835 (br sc) | 2.400 (s) | |||||
| Z-3a | 208-210 | 8.265(sc) | 8.14(dd) | 7.500-7.050 (m) | 4.836 (br s) | 2.400 (s) | |
| E,Z-3b | 250-253 | 8.158(se) | 8.168(df) | 7.713(df) | 7.352(br s) | 5.274 (br se) | --- |
| 8.275(se) | 4.870 (br se) | ||||||
| Z-3b | 188-190 | 8.274(de) | 8.144(de) | 7.831(br s) | 7.351 (br s) | 4.868 (br s) | --- |
| E,Z-3c[10] | 251-252 | 8.135(s) | 8.257(dg) | 7.686 (dg) | 7.353(br s) | 5.288 (br sc) | --- |
| 8.161(sc) | 5.050 (br sc) | ||||||
| Z-3c | 239-240 | 8.161(s) | 8.255(dg) | 7.700(dg) | 7.349(br s) | 5.050 (br s) | -- |
| Z-5a | 179-181 | 8.271(s) | 8.108(dg) | 7.433(dg) | 7.355(br s) | 4.862 (br s) | 2.420 (s) |
| Z-5b | 226-228 | 8.280(s) | 8.145(dg) | 7.832(dg) | 7.352(br s) | 4.869(br s) | --- |
| Z-5c | 226-228 | 8.291(s) | 8.234(df) | 7.689(df) | 7.352(br s) | 4.850 (br s) | |
- * Ha and Hb are the protons at the ortho- and meta positions of the aroyl group.
- a The spectrum shows also the following signals characteristic for AMX system: δ: 5.505 (dd, HA), 4.815 (dd,HM), 3.920(dd,HX), JHA,HM = 9.0 Hz, JHA,HX = 3.5 Hz, JHM,HX = 18.0 Hz.
- b J = 7.5 Hz.
- c Characteristic of E- and Z-isomers with integrated proton ratios of 15:1 respectively.
- d J = 7.2 Hz.
- e Characteristic of E- and Z-isomers with integrated proton ratios of 9:1, respectively;
- f J = 8.6 Hz.
- g J = 8.2 Hz.
| Compd. No. | IR (cm-1) C=O | EI-MS m/e(%) | Formula (M.W) | Analysis: Calcd/Found | ||||
| [M.+] | [A] | [B] | C | H | N | |||
| 2b | 1710,1690 | --- | --- | --- | C18H14BrNO2S2 | 51.43/ | 3.36/ | 3.33/ |
| (420.37) | 51.60 | 3.40 | 3.39 | |||||
| Z-3a | 1710,1680 | 353 | 309 | 204 | C19H15NO2S2 | 64.56/ | 4.27/ | 3.96/ |
| (5.1) | (39.3) | (23.9) | (353.47) | 64.43 | 4.25 | 4.03 | ||
| E,Z-3b | 1710,1680 | --- | --- | --- | C18H12BrNO2S2 | 51.68/ | 2.89/ | 3.35/ |
| (418.35) | 51.65 | 2.96 | 3.42 | |||||
| Z-3b | 1710,1680 | 417 | 373 | 268 | C18H12BrNO2S2 | 51.68/ | 2.89/ | 3.35/ |
| (13.7) | (15.8) | (12.0) | (418.35) | 51.73 | 3.02 | 3.50 | ||
| Z-3c | 1715,1690 | 373 | 329 | 224 | C18H12ClNO2S2 | 57.82/ | 3.23/ | 3.75/ |
| (3.0) | (35.7) | (22.2) | (373.89) | 57.91 | 3.15 | 5.62 | ||
| Z-5a | 1745,1690 | 337 | 309 | 204 | C19H15NO3S | 67.64/ | 4.48/ | 4.15/ |
| (2.0) | (39.3) | (23.9) | (337.40) | 67.73 | 4.39 | 4.06 | ||
| Z-5b | 1750,1690 | 401 | 373 | 268 | C18H12BrNO3S | 53.74/ | 3.01/ | 3.48/ |
| (3.7) | (22.0) | (12.2) | (402.29) | 53.711 | 3.10 | 3.52 | ||
| Z-5c | 1750,1690 | 357 | 329 | 224 | C18H12ClNO3S | 60.42/ | 3.38/ | 3.91/ |
| (2.1) | (35.7) | (22.2) | (357.83) | 60.56 | 3.49 | 3.83 | ||
References
- Omar, M.T.; Fouli, F.A.; El-Garhi, M.Z. Bull Chem. Soc. Jpn. 1991, 64, 750.
- Omar, M.T.; Kandeel, K.A.; Youssef, A.S.A. Monatsh. Chem. 1995, 126, 439.
- Omar, M.T.; Youssef, A.M. Org. Prep. and Proc. Int. 1991, 23(3), 379.
- Omar, M.T.; Youssef, A.M. Monatsh. Chem. 1991, 122, 263.
- Omar, M.T.; Youssef, A.M. Phosph. Sulfur and Silicon 1990, 35, 267.
- Omar, M.T.; Kasem, M.A. J. Heterocyclic Chem. 1981, 18, 1413.
- Kassab, N.A.; El-Nagdy, M.H.; Ead, H.A.R. J. Prakt. Chem. 1973, 315, 265.
- Ginak, A.I.; Sochilin, E.G. Zh. Org. Khim. 1978, 14, 1065.
- Hassaneen, H.M.; Shawali, A.S.; Farag, D.S.; Ahmed, E.M. Phosph. Sulfur and Silicon 1996, 113, 53.
- Nagase, H. Chem. Pharm. Bull. 1974, 22, 1661. [PubMed]
- Desarlo, F. J. Chem. Soc. Perkin I 1974, 1951.
- Pascual, C.; Meler, J.; Simon, W. Helv. Chim. Acta 1966, 49, 164.
- Firestone, R.A. Tetrahedron 1977, 33, 3009. Luich, J.M.; Bertran, J. Tetrahedron 1982, 38, 1847. Hiberty, P.C.; Ohanessian, J.; Schlegel, H.B. J. Amer. Chem. Soc. 1983, 105, 719.
- Huisgen, R. Angew. Chem., Int. Ed. Engl. 1963, 2, 565.
- Huisgen, R.; Mack, W.; Anneser, E. Angew. Chem. 1961, 73, 656.
- Huisgen, R.; Grashey, R.; Seidel, N.; Knupfer, H.; Schmidt, R. Liebigs. Ann. 1962, 658, 169.
- Papa, D.; Schwenk, E.; Villani, F.; Klingsberg, E. J. Amer. Chem. Soc. 1948, 70, 3356.
- Kinugawa, J.; Nagase, H. Japanese Patent 1964, 11,342 (66) (Cl.16E 351). Chem. Abst. 1966, 65, 13717d. [Google Scholar]
- Wiley, R.H.; Wakefield, B.J. J. Org. Chem. 1960, 25, 546.
- Kinney, C.R.; Smith, E.W.; Wolley, B.L.; Willey, A.R. J. Amer. Chem. Soc. 1933, 55, 3418.
- Samples Availability: Samples are available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Kandeel, K.A.; Youssef, A.S.A. Reactions of 5-Aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines with Nitrile Oxides. Molecules 2001, 6, 510-518. https://doi.org/10.3390/60600510
AMA Style
Kandeel KA, Youssef ASA. Reactions of 5-Aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines with Nitrile Oxides. Molecules. 2001; 6(6):510-518. https://doi.org/10.3390/60600510
Chicago/Turabian StyleKandeel, Kamal A., and Ahmed S. A. Youssef. 2001. "Reactions of 5-Aroylmethylene-3-benzyl-4-oxo-2-thioxo-1,3-thiazolidines with Nitrile Oxides" Molecules 6, no. 6: 510-518. https://doi.org/10.3390/60600510