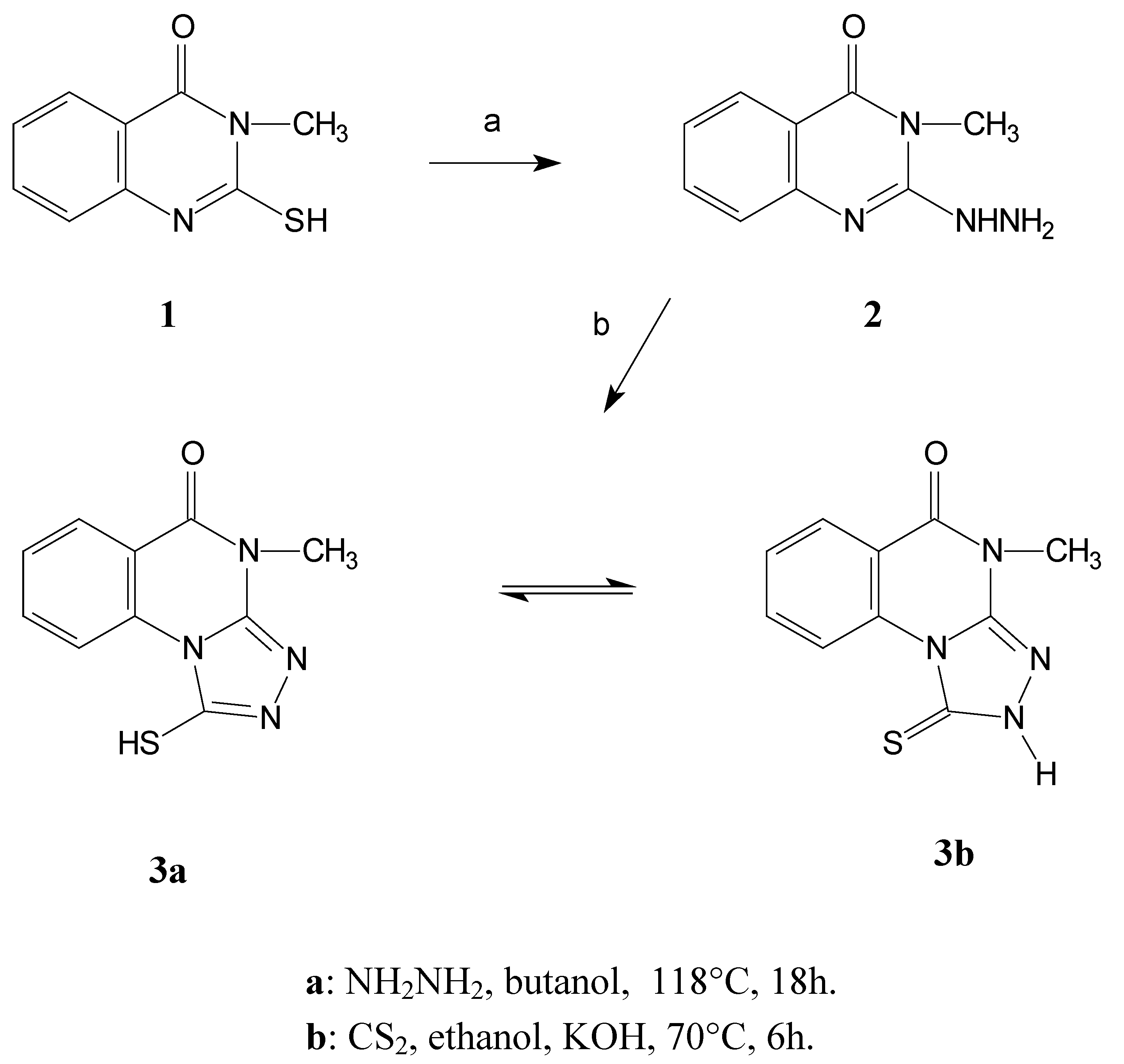
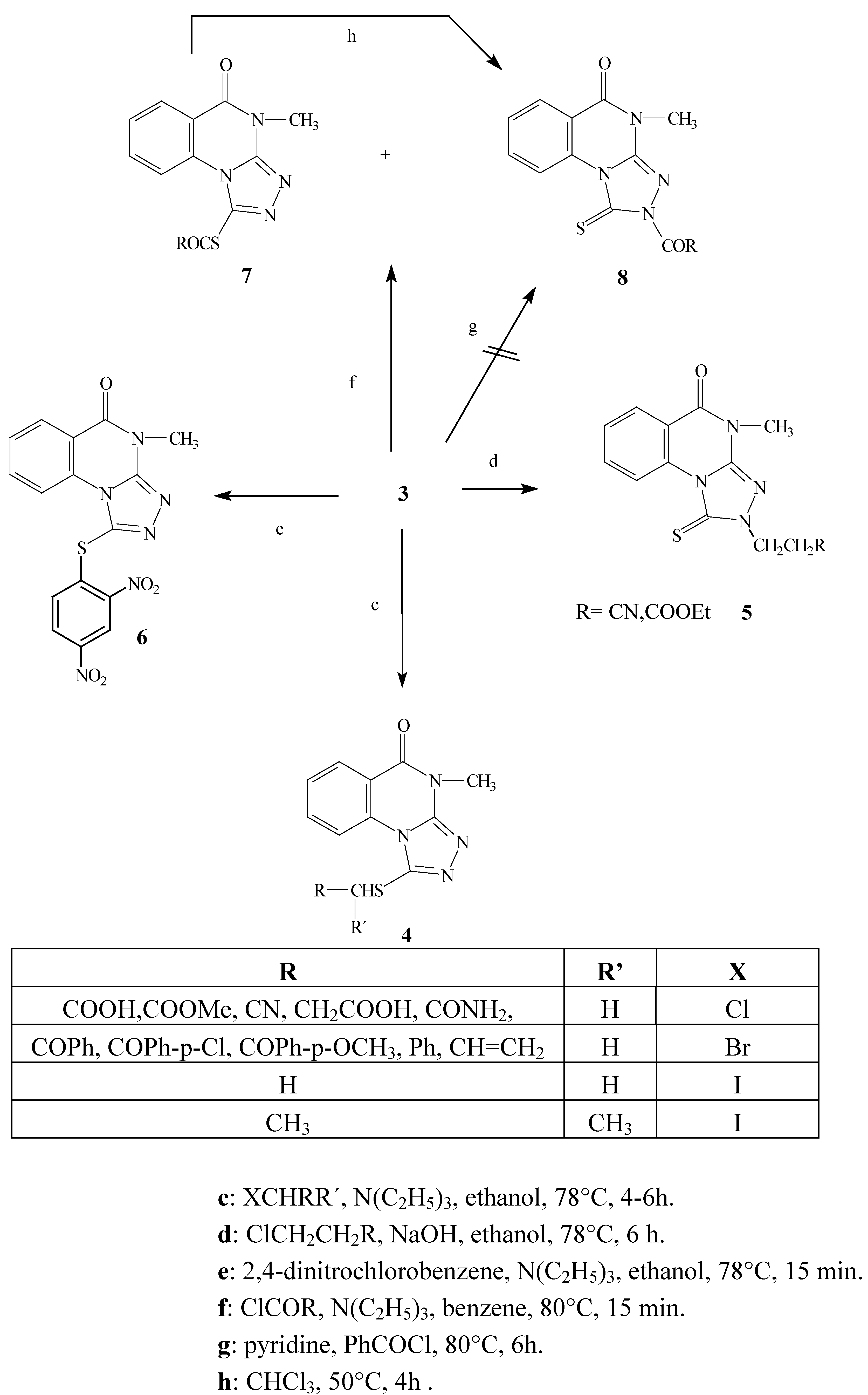
Results and Discussion
In this paper we report the results of a study of the regioselectivity of reactions of various electrophilic reagents (alkyl, acyl and activated aryl halides) with the thioamide moiety of the model compound 4-methyl-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (3).
The theoretical DFT studies of the anion formed by deprotonation of
3 (
Figure 1a and
Figure 1b; results are summarized in
Table 1,
Table 2 and
Table 3) show that the anionic form is completely planar thus conferring aromaticity to the whole system. On the other hand, the C(1) - N(2) bond order value would suggest its double bond character, as could be expected for form
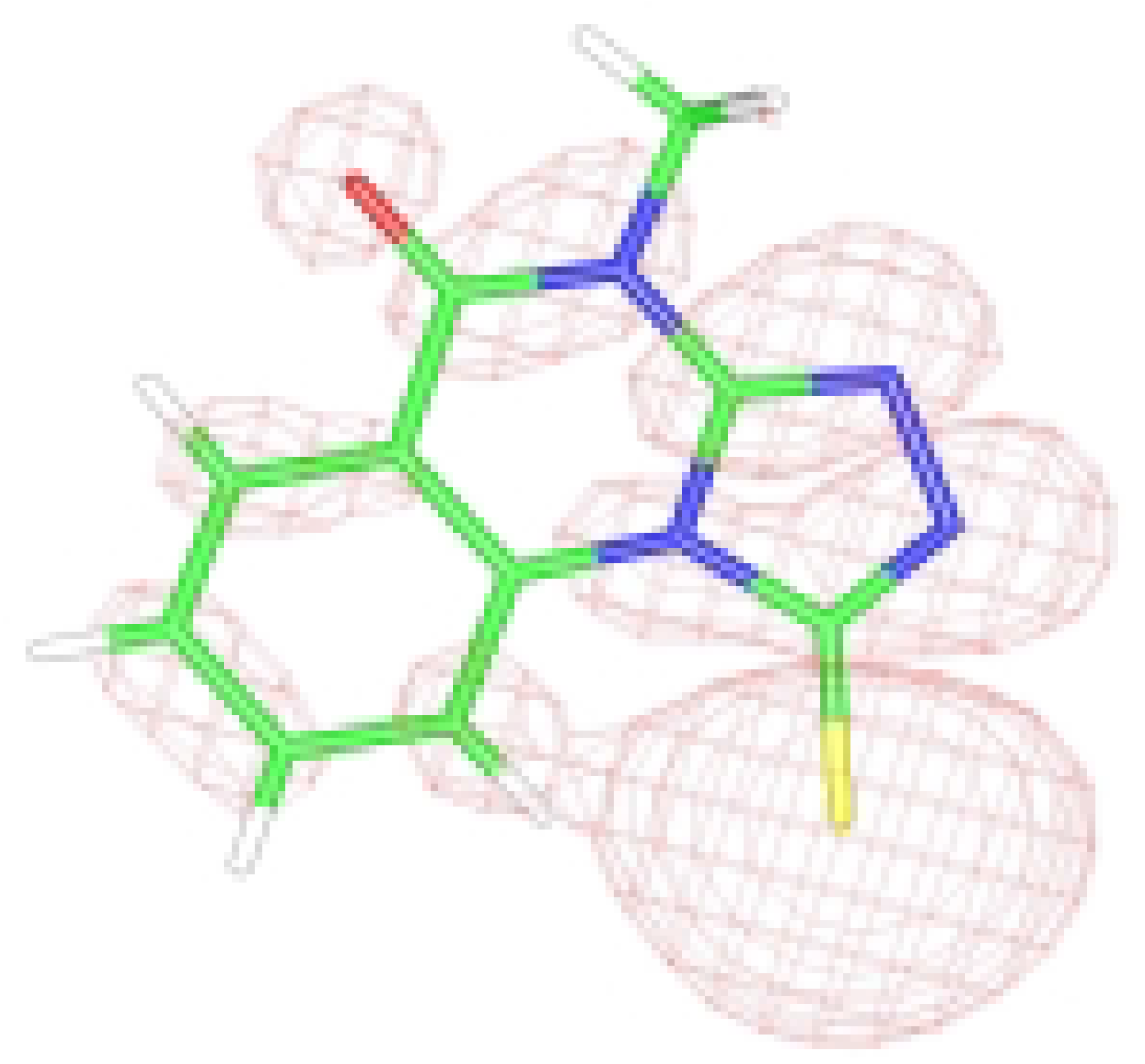
3a. However, the C(1) - S(11) bond order value is even larger, which indicates the delocalization of negative charge between the sulfur and nitrogen atoms. A residual bond order determined between the H(17) and S(11) atoms show the weak H-bond character of the interaction between them.
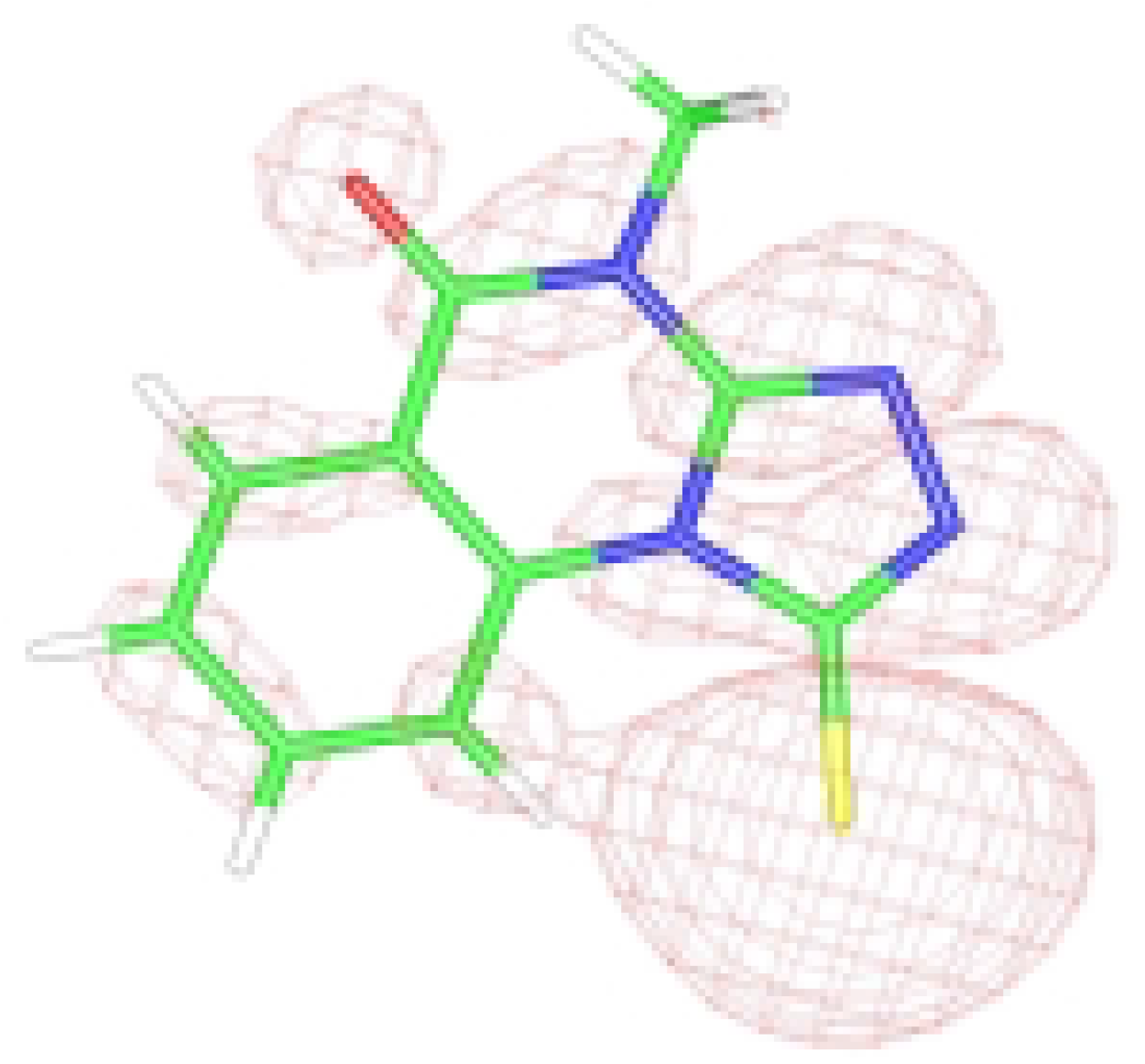
The results of the theoretical DFT studies gave a larger value for the HOMO coefficient c
2 on sulfur than the same on N(2). Finally, the partial charge distributed on the sulfur atom is slightly higher than that on the nitrogen atom [
6]. These results indicate that electrophilic attack on the sulfur atom might be preferred, in disagreement with literature data mentioned above.
These facts led us to focus our interest on controlled reactions involving only one of these two forms and to examine reaction of these two structural forms with different electrophiles. The problems faced are the elucidation of product structure if the reaction takes place either at the nitrogen or the sulfur atoms, or on both, the secondly, to give a satisfactory explanation to account for such a behavior.
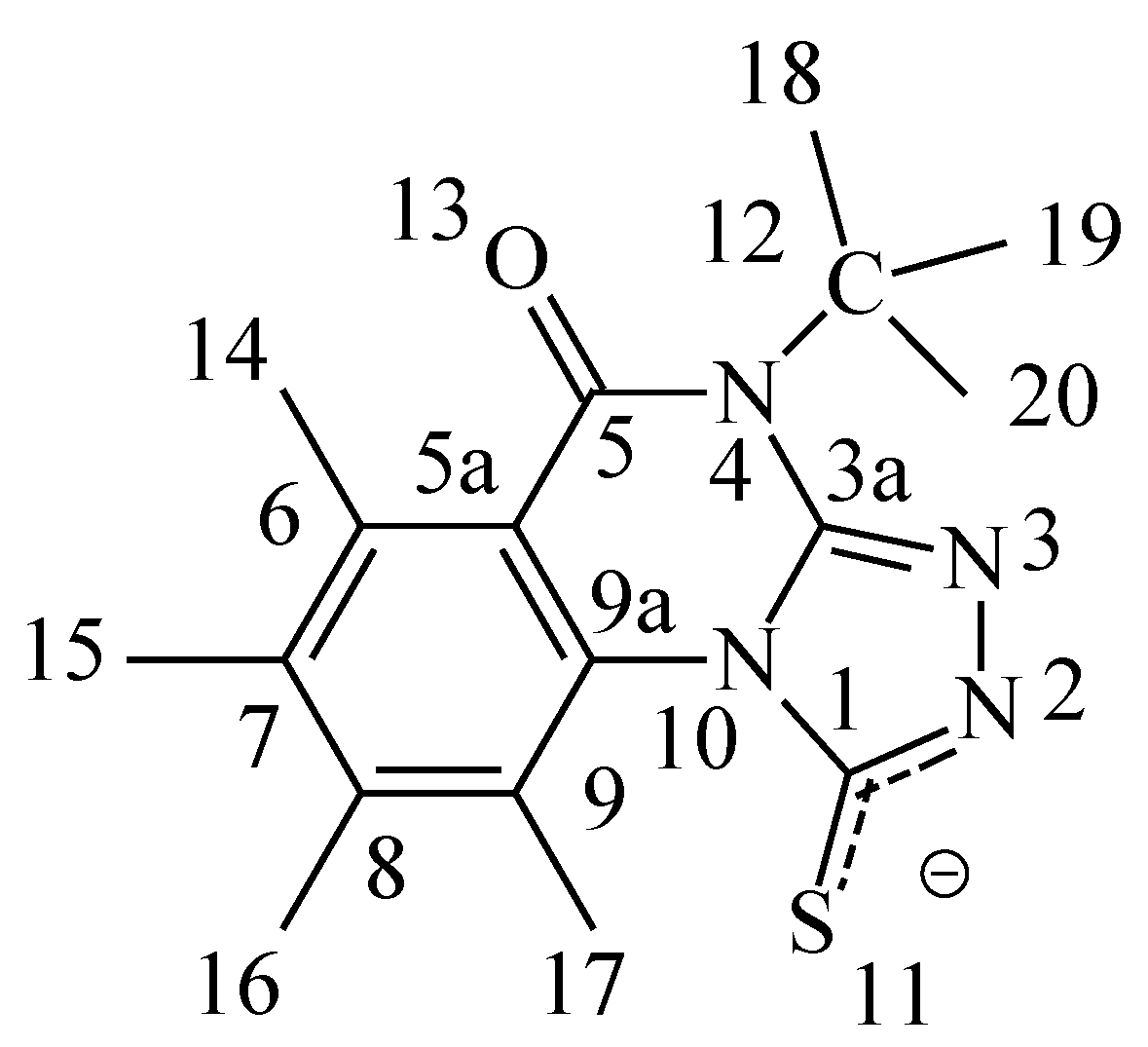
Figure 1a.
Numbering of the model compound 3 - anion.
Figure 1a.
Numbering of the model compound 3 - anion.
Figure 1b.
HOMO graphical representation of the anion form of 3.
Figure 1b.
HOMO graphical representation of the anion form of 3.
Table 1.
The molecular orbital coefficient values of the anion form of 3.
Table 1.
The molecular orbital coefficient values of the anion form of 3.
| HOMO | LUMO |
|---|
| atom | orbital | coefficient c | c2 | atom | orbital | coefficient c | c2 |
| N2 | 2pz | -0.2781 | 0.0740 | N2 | 2pz | -0.0812 | 0.0066 |
| C3a | 2pz | 0.1886 | 0.0355 | C3a | 2pz | 0.0239 | 0.0005 |
| N3 | 2pz | 0.1441 | 0.0207 | N3 | 2pz | 0.0300 | 0.0009 |
| N4 | 2pz | -0.1039 | 0.0108 | N4 | 2pz | -0.1971 | 0.0388 |
| C9 | 2pz | 0.0326 | 0.0010 | C9 | 2pz | -0.1562 | 0.0244 |
| C9a | 2pz | -0.0014 | 0 | C9a | 2pz | -0.2029 | 0.0411 |
| C5a | 2pz | 0.0300 | 0.0009 | C5a | 2pz | 0.2396 | 0.0574 |
| C6 | 2pz | 0.0333 | 0.0011 | C6 | 2pz | -0.2304 | 0.0531 |
| C5 | 2pz | -0.0552 | 0.0030 | C5 | 2pz | 0.2674 | 0.0715 |
| 8C | 2pz | -0.0322 | 0.0010 | 8C | 2pz | 0.2948 | 0.0869 |
| O13 | 2pz | 0.0715 | 0.0051 | O13 | 2pz | -0.2179 | 0.0475 |
| H17 | 1s | 0.0000 | 0 | H17 | 1s | -0.0000 | 0 |
| | 2s | -0.0000 | 0 | | 2s | -0.0000 | 0 |
| S11 | 2pz | -0.2070 | 0.0428 | S11 | 2pz | -0.0078 | 0 |
| 3pz | 0.5352 | 0.2864 | 3pz | 0.0230 | 0.0005 |
Table 2.
The partial charge values of selected atoms in the anion form of 3.
Table 2.
The partial charge values of selected atoms in the anion form of 3.
| | Mulliken | ESP | | Mulliken | ESP | | Mulliken | ESP |
| 1C | 0.2998 | 0.2615 | 5C | 0.5793 | 0.5440 | 10N | -0.6558 | -0.0286 |
| 2N | -0.3620 | -0.3966 | 5aC | 0.0077 | 0.0651 | 11S | -0.4943 | -0.6419 |
| 3N | -0.4398 | -0.4141 | 7C | -0.1357 | -0.2638 | 12C | -0.3134 | -0.1919 |
| 3aC | 0.7242 | 0.5434 | 8C | -0.1481 | -0.0242 | 13O | -0.5631 | -0.5354 |
| 4N | -0.5512 | -0.2757 | 9aC | 0.3345 | 0.1136 | 17H | 0.1947 | 0.1385 |
Table 3.
Bond orders of selected bonds in the anion form of 3.
Table 3.
Bond orders of selected bonds in the anion form of 3.
| S11-H17 | 0.1326 | N4-C3a | 1.0977 | C5-N4 | 1.1996 |
| C9a-N10 | 1.0707 | C3a-N10 | 1.2015 | C1-N2 | 1.6805 |
| C5a-C5 | 1.1157 | C3a-N3 | 1.8301 | C1-N10 | 0.9445 |
| C5-O13 | 1.8372 | N2-N3 | 1.0744 | C1-S11 | 2.0511 |
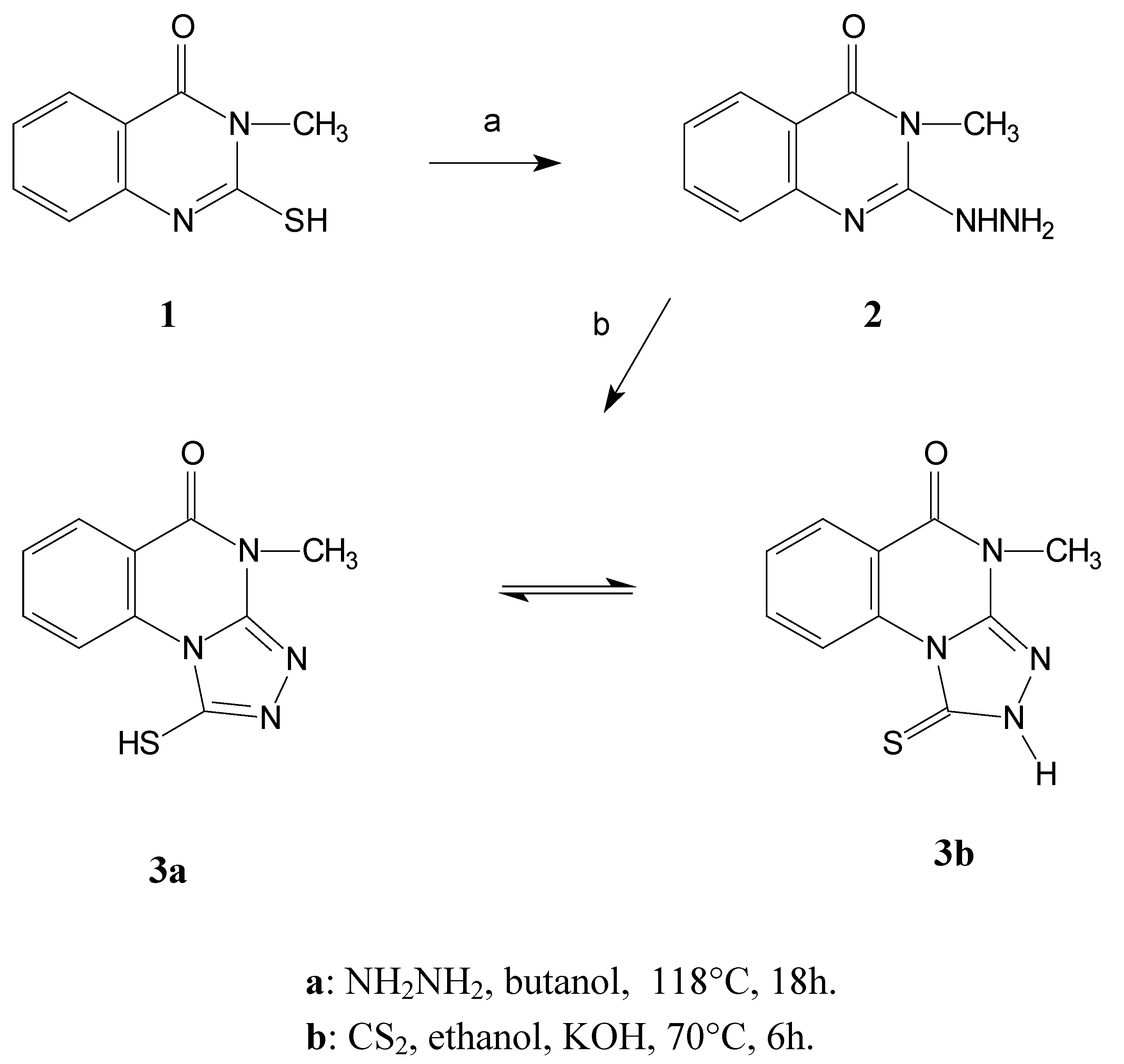
Compound
3 was prepared by a series of reactions starting from the easily available 3-methyl- 2-sulfanyl-3,4-dihydro-4-quinazolinone
1 [
7]. Hydrazinolysis of this starting quinazoline afforded the hydrazine derivative
2 that subsequently reacts with carbon disulfide to give the desired product
3 [
8].
The triazoloquinazoline derivative displays an interesting tautomeric equilibrium between the thiol (3a) and thione (3b) forms. This was proven by 1H-NMR spectroscopy ((CD3)2SO), which shows a mixture of these two forms in a ratio of about 1:1 thiol 3a to thione 3b forms.
Scheme 1.
Synthesis of the model compound 3
Scheme 1.
Synthesis of the model compound 3
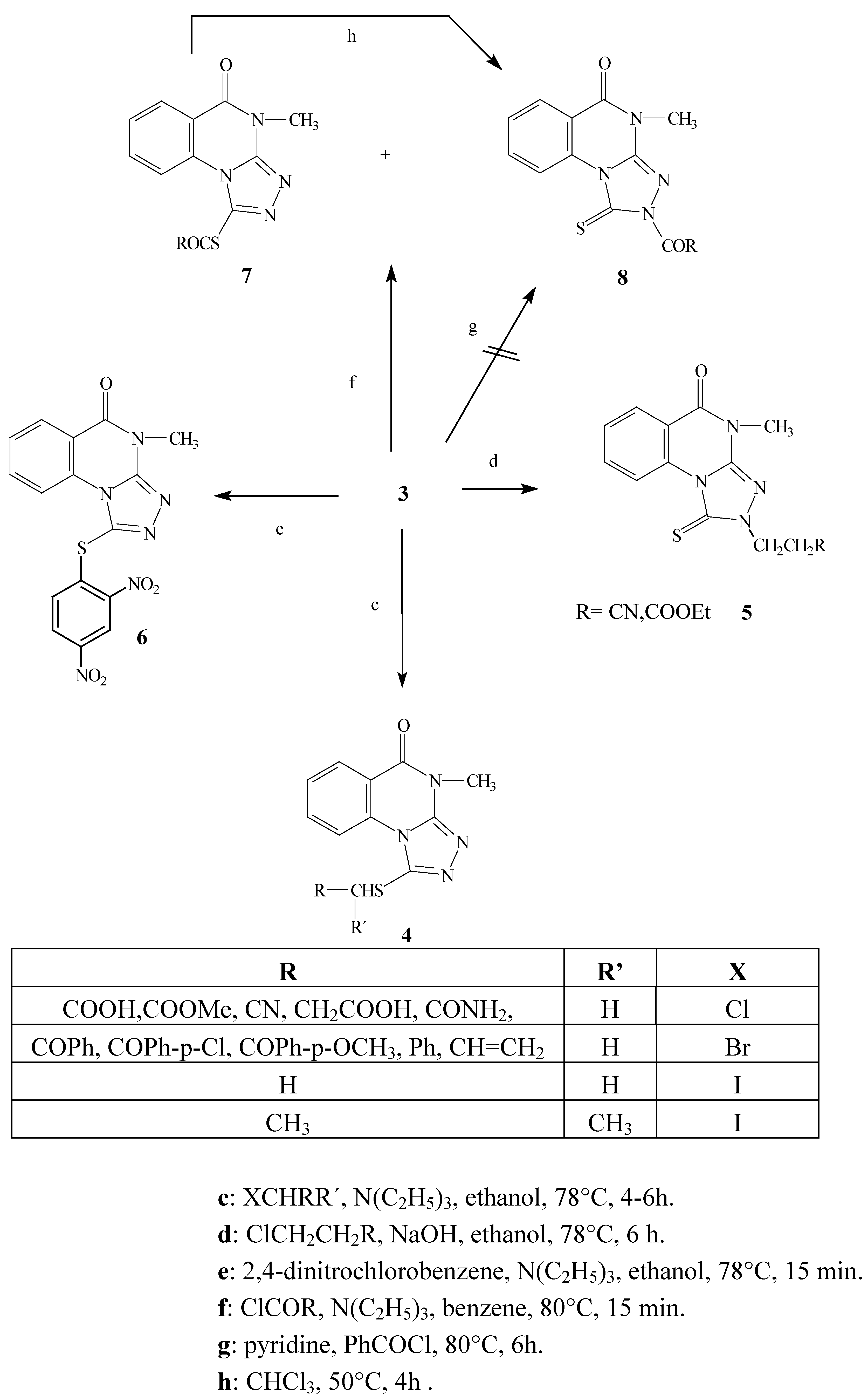
Scheme 2.
Alkylation, acylation and arylation reactions of the model compound 3.
Scheme 2.
Alkylation, acylation and arylation reactions of the model compound 3.
The ambident nucleophile 3 reacts with alkyl halides in several solvents (absolute ethyl alcohol, 95% ethyl alcohol and benzene) under basic conditions (K2CO3, NEt3 to produce the S-substituted products 4 via an SN2 reaction mechanism with almost all the alkyl halides used. On the other hand, when either 3-chloropropanenitrile and methyl-3-chloropropanoate were used, the reaction gave the N(2)-substituted products 5.
We might expect that the reaction would take place on the softer side of the ambident nucleophile (the sulfur atom) because of the soft character of the alkyl halides [
9]. Indeed, the reactions of alkyl halides with triazoloquinazoline in ethanol and triethylamine generally took place at the sulfur atom. The reaction is can be explained by an interaction between the HOMO of the nucleophile and the LUMO of the electrophile to produce the
S-attack. The reaction involves the attack of the nucleophile at the carbon atom of the electrophile to give the S
N2 transition state (the carbon atom is coordinated to three substituents and partially bonded to both the leaving group (halogen atom) and the ambident nucleophile, then the halide as a leaving group is expelled and the carbon atom regains tetra coordination. The ordering of S
N2 transition state inhibits the attack at the nitrogen atom due to the steric effects caused the bulky rigid part of the planar triazole side.
The alternative pathway is the formation of this transition state at the sulfur atom, which has several advantages compared to the former nitrogen attack. The sulfur atom attack is preferred due to sterically free access, higher partial charge and better polarizability due to its 3p orbital.
Several attempts were made to carry out the reaction of 3-chloropropanenitrile and methyl-3- chloropropanoate with
3 under different reaction conditions. It was found the reaction only took place in ethyl alcohol and in the presence of sodium hydroxide as a base to produce the
N(2)- substituted alkyl derivatives
5. The reagents 3-chloropropanenitrile and methyl-3-chloropropanoate underwent an elimination reaction to give acrylonitrile or methyl acrylate
in situ, respectively. The presence of the planar C=C double bond conjugated with the electron withdrawing nitrile or methoxycarbonyl group produced an electron deficiency on the C3 carbon atom, consequently giving a partial positive charge on this atom. Interaction between both planar acrylic and triazole systems is free of steric hindrance, too. On the other hand, the energy of LUMO in both acrylic acid functional derivatives is decreased and the coupling of activated C=C double bond takes place at the nitrogen atom. The reaction is produced by strong coulombic attraction at the hard part of the ambident nucleophile to finally produce the
N-substituted alkyl derivatives
5. The same products
5 are formed when the ambident nucleophile reacts with acrylonitrile and methyl acrylate. This gave more evidence for the involvement of methyl acrylate and acrylonitrile in the course of the reaction [
6].
The reaction of
3 with 3-chloropropanoic acid might show some similarity to the proceeding reactions, since we might expect the elimination of hydrogen chloride followed by reaction with
3 to give the
N-alkyl derivative. Our result was completely different in this case, yielding the
S-alkyl derivative
4. The same result was obtained 3-bromopropanoic acids were used. This is attributed to the weak electron withdrawing ability of the carboxylate anion compared to both the nitrile and ester group in the former reaction. The previously mentioned factor facilitates the elimination reaction in the case of 3-chloropropanenitrile and methyl-3-chloropropanoate, and is not effective in the case of the salts of both 3-chloro- and 3-bromopropanoic acid. It is noteworthy that acrylic acid gave no reaction with
3 under various reaction conditions [
6], which indicates that alkylation with 3-chloropropanoic acids only leads to the
S-substituted alkyl derivatives.
The reaction of the triazolo derivative 3 with the 2,4-dinitrochlorobenzene in the presence of triethylamine gave the S-aryl product 6 by a nucleophilic aromatic substitution reaction via formation of Meisenheimer’s complex. As described the reaction seems to be similar to the SN2 reaction and this explains the formation of the S-substituted aryl derivative.
The reaction of triazoloquinazoline 3 with pivaloyl chloride or ethyl chloroformate gave a mixture of the S- (minor) 7 and N-acyl (major) 8 products after 15 min reflux. Extending the reaction time resulted in the formation of only the N-acyl product 8. On the other hand the reaction of 3 with benzoyl chloride gave only the N-acyl derivative 8. It is noteworthy that the acylation reaction using the acyl halides in the presence of pyridine as a solvent and base gave an acyl pyridinium cation which is known as a hard electrophile, so applying this reaction condition to our ambident nucleophile and the acyl halides might lead to the sole formation of the N-acyl derivative 8. In fact no result was observed under the former reaction conditions, which confirms that the N- acyl derivative is not directly formed due to steric effects. We conclude that the reaction of the acyl halides in presence of benzene and triethylamine is a kinetically controlled reaction and results in formation of the S-acyl product 7 via orbital-interactions. This product undergoes a trans-acylation reaction to give the thermodynamically controlled N-acyl derivative 8 under the effect of prolonged reaction times or by heating the reaction mixtures. The trans-acylation reaction is proceeded by the formation of the acyl carbocation formed under elevated temperature by the AB1 mechanism, and is capable of attacking the nitrogen atom to produce the N-substituted acyl derivative.
The elucidation of N- and S-substituted derivative structures was performed using several analytical techniques: the IR spectra generally indicate the absence of the ν(ΝΗ) of compounds 3a- b and also show the characteristic frequency of the new-formed functional group.
The mass spectra of all examined compounds showed the molecular ion. It is apparent that the mass spectra do not give reliable evidence to discriminate between the S- and N- substituted derivatives.
1H-NMR spectroscopy did provide substantiation for the structures proposed for the
N- and
S-products, respectively. Thus, the
1H-NMR spectra displayed an interesting multiplet peak corresponding to H(17) at chemical shifts ranging from ca. 10.50-10.26 ppm in the case of both the N-alkyl (
5) or N-acyl (
8) derivatives [
6]. The high chemical shift is attributed to magnetic anisotropy interaction between C=S(11)….H(17) as shown in
Figure 1. This peak was used as an indicator for the
N-substituted derivatives; the corresponding peaks for H(17) in the case of the
S-substituted derivatives occurs at chemical shifts ranging from ca. 8.66-8.41 ppm for all examined compounds
4a-
l. The
1H-NMR and
13C-NMR chemical shifts for the SCH
2 group of the S-alkylated derivatives
4b-
l are shown in
Table 4.
Table 4.
1H NMR and 13C NMR chemical shifts for the SCH2 group of the S-alkylated derivatives 4b-l
Table 4.
1H NMR and 13C NMR chemical shifts for the SCH2 group of the S-alkylated derivatives 4b-l
| Compound no. | SCH2R | solvent | 1H-NMR chemical shift (SCH2) ppm | 13C-NMR chemical shift (SCH2) ppm |
|---|
| 4b | SCH2COOH | DMSO-d6 | 4.18 | 36.69 |
| 4c | SCH2COOCH3 | CDCl3 | 4.23 | 35.92 |
| 4d | SCH2CONH2 | DMSO-d6 | 4.04 | 37.50 |
| 4e | SCH2COPh | CDCl3 | 5.08 | 42.82 |
| 4f | SCH2COPh-p-Cl | CDCl3 | 5.02 | 42.33 |
| 4g | SCH2COPh-p-OCH3 | CDCl3 | 5.03 | 42.71 |
| 4h | SCH2Ph | CDCl3 | 4.63 | 38.94 |
| 4i | SCH2CH=CH2 | CDCl3 | 4.15 | 36.79 |
| 4j | SCH2CN | CDCl3 | 4.24 | - |
| 4k | SCH2CH2COOH | DMSO-d6 | 3.42 | 33.59 |
The 1H-NMR spectra of the N(2) substituted derivatives 5a and 5b gave peaks at chemical shifts of ca 4.59 ppm, corresponding to the NCH2 groups, while the 13C-NMR spectra show peaks at chemical shifts of ca 44.32 and 44.14 ppm, respectively. It is apparent from the reported data that the 1H-NMR and 13C-NMR chemical shifts of the SCH2 groupings occur at lower chemical shift than those of NCH2 taking into consideration the environment for the compared structures and the solvents used for the NMR experiments. Finally, the 13C-NMR spectra show peaks at chemical shifts of ca 163 ppm due to the C=S moiety which is associated with the N-acyl (8) and N-alkyl (5) derivatives, while S-alkylation gave peaks at a chemical shift of ca 144 ppm for the same carbon C(1) (corresponding to C-S).
Experimental
General
Melting points of all the compounds were measured on a Boetius Rapido PHMK 79/2106 (Wägetechnik) instrument. TLC was carried out on Silufol UV 254 plates (Kavalier, Votice); the eluent used was a 1: 4 mixture of acetone and benzene. TLC detection was accomplished with a Fluotes Universal (Quarzlampen, Hanau) instrument and iodine vapours. The purity of compounds 2-8 was verified by their elemental analyses, measured with an 1102 (Erba) instrument. FTIR spectra were taken on a Genesis (Unicam) spectrometer using potassium bromide pellets. NMR spectra were measured on a Bruker Avance DRX-500 spectrometer. 1H- and 13C-NMR spectra were measured in CDCl3, (CD3)2SO or CF3COOD solutions and tetramethylsilane was used as an internal standard. The measured 13C- and 1H- NMR spectra were correlated with those obtained by on-line simulation (Advanced Chemistry Development, Inc., Toronto, Canada). Mass spectra measured (electron impact, 70 eV) were with a FISONS INSTUMENTS TRIO 1000 and GC 8000 series apparatus.
The theoretical results were obtained using the DFT method at the B3LYP/6-31G** level [
10,
11]. Mulliken [
12], ESP [
13] and NBO [
14,
15,
16] methods were used to calculate the partial charges and properties of molecular orbitals. Mayer bond orders [
17] were calculated at a B3LYP/ii-iglo level using the demon v.l program [
18].
The starting materials
1,
2 and
3 were prepared according to the method reported in literature [
8].
2-Hydrazino-3-methyl-3,4-dihydro-4-quinazolinone (2).
A mixture of quinazoline derivative 1 (1.92 g, 0.01 mol) and hydrazine hydrate (2.5 ml, 0.05 mol) in butanol (40 ml) was stirred under reflux for 18 h until the evolution of hydrogen sulfide ceased. The reaction mixture was allowed to cool, and the resultant solid was filtered off. After dissolution of the product thus obtained in 3M hydrochloric acid solution, it was reprecipitated by the addition of aqueous ammonia solution to pH 8, filtered, washed with water and crystallized from ethyl alcohol to give 1.42 g (65%) of the title compound as white crystals; M.p. 209-210°C; Calculated for C9H10N4O (190.21): 56.84% C, 5.26% H, 29.47% N; Found: 56.25% C, 5.01% H, 29.20% N; FTIR, ṽ /cm-1: 3300, 3268 (NH2), 1661 (lactam C=O).
4-Methyl-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (3).
To a suspension of hydrazine derivative 2 (1.9 g, 0.01 mol) in ethyl alcohol, a solution of potassium hydroxide (1.12 g, 0.02 mol) in water (10 mL) was added. After the addition of carbon disulfide (7.6 mL, 0.1 mol) the reaction mixture was refluxed gently on a water bath at 70 °C for 6 h. Afterwards the solvent was evaporated under reduced pressure to complete dryness. The residue was redissolved in water and the solution thus obtained was acidified with 1N hydrochloric acid solution to pH 1. The resultant flocculent precipitate was filtered, washed with water and crystallized from DMF to afford 1.74 g (75%) of the desired product as yellowish crystals M.p. 285-286°C; Calculated for C10H8N4OS (232.27): 51.72% C, 3.45% H, 24.14% N, 13.80% S, Found: 52.00% C, 3.81% H, 24.32% N, 13.65% S; FTIR, ṽ /cm-1: 3177 (NH), 1672 (C=O, cyclic amide). The 1H-NMR proves the presence of the tautomeric mixture of 3a and 3b in about a 1:1 ratio. 3a: 1H-NMR (DMSO-d6) δ/ ppm: 8.22-8.20 (1H, m, ArH), 7.90-7.85 (1H, m, ArH), 7.62-7.57 (1H, m, ArH), 7.40-7.37 (1H, m, ArH), 3.65 (3H, s, NCH3); Second series of chemical shifts corresponding to 3b: 10.21-10.18 (1H, m, ArH), 7.97-7.94 (1H, m, ArH), 7.74-7.69 (1H, m, ArH), 7.34-7.30 (1H, m, ArH), 3.44 (3H, s, NCH3). 1H-NMR (CF3COOD) δ/ ppm: 10.44-10.41 (1H, m, ArH), 8.87-8.85 (1H, m, ArH), 8.40-8.35 (1H, m, ArH), 8.15-8.10 (1H, m, ArH), 4.19 (3H, s, NCH3). 13C-NMR (CF3COOD) δ/ ppm: 164.12 (C=S), 163.64 (lactam C=O), 148.69 (Cq), 138.17 (CHAr), 137.00 (Cq), 131.67 (CHAr), 131.03 (CHAr), 119.21 (CHAr), 31.44 (NCH3).
General Procedure for S-Alkylation with Alkyl Halides
To a mixture of the triazolo derivative 3 (2.32 g, 0.01 mol) and triethylamine (2 mL, 0.02 mol) in ethyl alcohol (30 mL) the appropriate alkyl halide (0.01 mol) was added. The reaction mixture was heated under reflux for 4-6 h, and then concentrated under reduced pressure. The solid obtained was filtered and then recrystallized from ethyl alcohol.
4-Methyl-1-(methylsulfanyl)-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (4a).
(Alkylating agent: methyl iodide): yield 1.55 g (63%); M.p. 234-235 °C; Calculated for C11H10N4OS (246.29): 53.65% C, 4.09% H, 22.75% N, 13.02% S, Found: 53.54% C, 3.98% H, 22.62% N, 12.77% S; FTIR, ṽ /cm-1: 1686 (C=O), 3000, 2922 (CH), 1613 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.45-8.42 (1H, m, ArH), 8.35-8.32 (1H, m, ArH), 7.85-7.80 (1H, m, ArH), 7.59-7.53 (1H, m, ArH), 3.80 (3H, s, NCH3), 2.90 (3H, s, SCH3); 13C-NMR (CDCl3) δ/ ppm: 158.74 (lactam C=O), 150.03 (Cq), 145.81 (C-S), 134.71 (CHAr), 133.74 (Cq), 130.24 (CHAr), 126.97 (CHAr), 117.44 (Cq), 115.99 (CHAr), 29.78 (NCH3), 16.27 (SCH3).
2-[(4-Methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl)sulfanyl]acetic acid (4b).
(Alkylating agent: chloroacetic acid); yield 1.89 g (65%); M.p. 205-206 °C; Calculated for C12H10N4O3S (290.30): 49.65% C, 3.47% H, 19.30% N, 11.04% S, Found: 49.42% C, 3.45% H, 19.25% N, 11.02% S; FTIR, ṽ /cm-1: 1675(C=O), 1711 (acid C=O), 3304-2908 (broad, acid-OH), 2987, 2955 (CH), 1616 (C=N); 1H-NMR (DMSO-d6) δ/ ppm: 8.41-8.39 (1H, m, ArH), 8.28-8.26 (1H, m, ArH), 7.99-7.96 (1H, m, ArH), 7.65-7.62 (1H, m, ArH), 4.18 (2H, s, SCH2CO), 3.58 (3H, s, NCH3); 13C-NMR (DMSO-d6) δ/ ppm: 169.86 (acid C=O), 158.95 (lactam C=O), 150.49 (Cq), 143.59 (C-S), 135.63 (CHAr), 134.07 (Cq), 129.89 (CHAr), 127.65 (CHAr), 117.92 (Cq), 116.52 (CHAr), 36.69 (SCH2), 29.95 (NCH3).
Methyl-2-[(4-methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl)sulfanyl]acetate (4c).
(Alkylating agent: methyl chloroacetate): yield 1.37 g (45%); M.p. 150-151 °C; Calculated for C13H12N4O3S (304.32): 51.31% C, 3.97% H, 18.41% N, 10.53% S, Found: 51.28% C, 3.83% H, 18.23% N, 10.51% S; ṽ /cm-1: 1675 (C=O), 1741 (ester C=O), 2987, 2955 (CH), 1616 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.45-8.41 (1H, m, ArH), 8.40-8.37 (1H, m, ArH), 7.85-7.80 (1H, m, ArH), 7.59-7.54 (1H, m, ArH), 4.23 (2H, s, SCH2CO), 3.78 (3H, s, OCH3), 3.76 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 168.51 (ester C=O), 158.71 (lactam C=O), 150.17 (Cq), 143.39 (C-S), 134.87 (CHAr), 133.59 (Cq), 130.51 (CHAr), 127.45 (CHAr), 117.52 (Cq), 116.54 (CHAr), 53.24 (OCH2), 35.92 (SCH2), 29.88 (NCH3); Mass spectrum, m/z (Ir/%): 304 (46), 273 (9), 246 (14), 245 (100), 244 (36), 231 (8), 203 (10), 162 (57), 159(11), 134 (24), 104 (4), 102 (7), 90 (28) , 76 (7).
2-[(4-Methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl) sulfanyl]acetamide (4d)
(Alkylating agent: chloroacetamide): yield 2.02 g (70%); M.p. 225-226 °C; Calculated for C12H11N5O2S (289.31): 49.82% C, 3.83% H, 24.21% N, 11.08% S, Found: 49.77% C, 3.62% H, 24.06% N, 10.83% S; ṽ /cm-1: 1690 (C=O), 1690 (amide C=O), 3355, 3173 (NH2), 3011, 2966, 2924 (CH), 1610 (C=N); 1H-NMR (DMSO-d6) δ/ ppm: 8.48-8.45 (1H, m, ArH), 8.26-8.23 (1H, m, ArH), 7.99-7.93 (1H, m, ArH), 7.68 (1H, bs, NH2), 7.64-7.59 (1H, m, ArH), 7.25 (1H, bs, NH2), 4.04 (2H, s, SCH2CO), 3.58 (3H, s, NCH3); 13C-NMR (DMSO-d6) δ/ ppm: 168.31 (amide C=O), 158.16 (lactam C=O), 149.57 (Cq), 143.07 (C-S), 134.79 (CHAr), 133.32 (Cq), 128.97 (CHAr), 126.79 (CHAr), 117.01 (Cq), 115.76 (CHAr), 37.50 (SCH2), 29.12 (NCH3).
4-Methyl-1-[(2-oxo-2-phenylethyl)sulfanyl]-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (4e).
(Alkylating agent: phenacyl bromide): yield 2.31 g (66%); M.p. 218-219 °C; Calculated for C18H14N4O2S (350.39): 61.70% C, 4.03% H, 15.99% N, 9.15% S, Found: 61.52% C, 3.89% H, 15.73% N, 9.08% S; ṽ /cm-1: 1682 (C=O), 1695 (C=O), 3039, 2974, 2917 (CH), 1611 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.46-8.45 (1H, m, ArH), 8.43-8.42 (1H, m, ArH), 8.04-8.03 (2H, m, ArH), 7.85-7.82 (1H, m, ArH), 7.64-7.61 (1H, m, ArH), 7.58-7.55 (1H, m, ArH), 7.51-7.48 (2H, m, ArH), 5.08 (2H, s, SCH2CO), 3.77 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 192.94 (C=O), 158.73 (lactam C=O), 150.05 (Cq), 144.25 (C-S), 135.35 (Cq), 134.87 (CHAr), 134.32 (CHAr), 133.60 (Cq), 130.26 (CHAr), 129.11 (CHAr), 128.78 (CHAr), 127.22 (CHAr), 117.45 (Cq), 116.32 (CHAr), 42.82 (SCH2), 29.79 (NCH3).
1-{[(2-(4-Chlorophenyl)-2-oxoethyl]sulfanyl}-4-methyl-4,5-dihydro[1,2,4]triazolo[4,3-a]- quinazolin-5-one (4f).
(Alkylating agent: 4-chlorophenacyl bromide): yield: 2.76 g (72%), M.p. 216-217 °C; Calculated for C18H13N4O2S (384.84): 56.18% C, 3.40% H, 9.21% Cl, 14.56% N, 8.33% S, Found: 56.16% C, 3.37% H, 9.11% Cl, 14.49% N, 8.33% S; ṽ /cm-1: 1680 (C=O), 1694 (C=O), 2970, 2907 (CH), 1615 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.45-8.44 (1H, m, ArH), 8.43-8.41 (1H, m, ArH), 8.00-7.97 (2H, m, ArH), 7.86-7.81 (1H, m, ArH), 7.60-7.55 (1H, m, ArH), 7.49-7.46 (2H, m, ArH), 5.02 (2H, s, SCH2CO), 3.77 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 191.86 (C=O), 158.73 (lactam C=O), 150.05 (Cq), 144.25 (C-S), 140.99 (CHAr), 134.91 (Cq), 134.87 (CHAr), 133.77 (Cq), 130.37 (CHAr), 130.20 (CHAr), 129.50 (CHAr), 127.31 (CHAr), 117.53 (Cq), 116.29 (CHAr), 42.33 (SCH2), 29.83 (NCH3).
1-{[(2-(4-Methoxyphenyl)-2-oxoethyl]sulfanyl}-4-Methyl4,5-dihydro[1,2,4]triazolo[4,3-a]- quinazolin-5-one (4g).
(Alkylating agent: 4-methoxyphenacyl bromide): yield 2.47 g (65%); M.p. 194-195 °C; Calculated for C19H16N4O3S (380.42): 59.99% C, 4.24% H, 14.73% N, 8.43% S, Found: 59.64% C, 4.21% H, 14.67% N, 8.36% S; ṽ /cm-1: 1670 (C=O), 1688 (C=O), 3009, 2959 (CH), 1614 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.49-8.47 (1H, m, ArH), 8.43-8.41 (1H, m, ArH), 8.02-7.99 (2H, m, ArH), 7.85-7.80 (1H, m, ArH), 7.58-7.54 (1H, m, ArH), 6.69-6.93 (2H, m, ArH), 5.03 (2H, s, SCH2CO), 3.88 (3H, s, OCH3), 3.77 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 191.43 (C=O), 158.78 (lactam C=O), 151.67 (Cq), 144.25 (C-S), 134.91 (Cq), 134.89 (CHAr), 133.67 (Cq), 131.21 (CHAr), 130.23 (CHAr), 128.38 (Cq), 127.22 (CHAr), 117.48 (Cq), 116.42 (CHAr), 114.31(CHAr), 55.79 (OCH3), 42.71 (SCH2), 29.80 (NCH3).
1-(Benzylsulfanyl)-4-methyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (4h).
(Alkylating agent: benzyl bromide): yield 2.13 g (66%); M.p. 151-152 °C; Calculated for C17H14N4OS (322.38): 63.34% C, 4.38% H, 17.38% N, 9.94% S, Found: 63.25% C, 4.36% H, 17.19% N, 9.81% S; ṽ /cm-1: 1679 (C=O), 3034, 2937 (CH), 1603 (C=N); 1H-NMR (CDCl3) δ/ppm: 8.43-8.39 (1H, m, ArH), 8.37-8.36 (1H, m, ArH), 7.77-7.71 (1H, m, ArH), 7.54-7.48 (1H, m, ArH), 7.44-7.42 (2H, m, ArH), 7.32-7.25 (3H, m, ArH), 4.63 (2H, s, SCH2Ph), 3.78 (3H, s, NCH3); 13C- NMR (CDCl3) δ/ ppm: 158.77 (lactam C=O), 149.94 (Cq), 144.58 (C-S), 135.84 (Cq), 134.67 (CHAr), 133.75 (Cq), 130.20 (CHAr), 129.53 (CHAr), 128.95 (CHAr), 128.26 (CHAr), 127.04 (CHAr), 117.45 (Cq), 116.26 (CHAr), 38.94 (SCH2), 29.80 (NCH3).
1-(Allylsulfanyl)-4-methyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (4i).
(Alkylating agent: allyl bromide): yield 1.50 g (55%); M.p. 110-111 °C; Calculated for C13H12N4OS (272.32): 57.34% C, 4.44% H, 20.57% N, 11.77% S, Found: 57.18% C, 4.38% H, 20.48% N, 11.53% S; ṽ /cm-1: 1681 (C=O), 2987, 2955 (CH), 1630 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.53-8.49 (1H, m, ArH), 8.31-8.27 (1H, m, ArH), 7.82-7.78 (1H, m, ArH), 7.64-7.62 (1H, m, ArH), 6.02-6.12 (1H, m, CH=CH2), 5.22 (1H, d, CH=CH2, J= 9.52 Hz), 5.09 (1H, d, CH=CH2, J= 8.4 Hz), 4.15 (2H, d, SCH2, J= 5.45 Hz) 3.80 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 158.58 (C=O, cyclic amide), 149.72 (Cq), 144.04 (C-S), 134.52 (CHAr), 133.61 (Cq), 131.86 (CH=CH2), 130.01 (CHAr), 129.89 (CHAr), 126.89 (CHAr), 119.89 (CH=CH2), 117.27 (Cq), 116.00 (CHAr), 36.79 (SCH2), 29.56 (NCH3); Mass spectrum, m/z (Ir/%): 272 (29), 257 (100), 239 (12), 231 (3), 203 (6), 162 (31), 159 (10), 143 (5), 134(14), 90 (19) , 76 (5).
2-[(4-Methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl)sulfanyl]acetonitrile (4j).
(Alkylating agent: chloroacetonitrile): yield 1.60 g (59%), M.p. 236-237 °C; Calculated for C12H9N5OS (271.30): 53.13% C, 3.34% H, 25.81% N, 11.82% S, Found: 53.11% C, 3.34% H, 25.79% N, 11.82% S; ṽ /cm-1: 1685 (C=O), 2248 (CN), 3048, 2980, 2933 (CH), 1610 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.50-8.48 (1H, m, ArH), 8.27-8.24 (1H, m, ArH), 7.90-7.85 (1H, m, ArH), 7.65-7.60 (1H, m, ArH), 4.24 (2H, s, SCH2), 3.81 (3H, s, NCH3); 1H NMR (CF3COOD) δ/ ppm: 8.80-8.77 (1H, m, ArH), 8.54-8.51 (1H, m, ArH), 8.34-8.29 (1H, m, ArH), 8.11-8.06 (1H, m, ArH), 4.62 (2H, s, SCH2), 4.12 (3H, s, NCH3); 13C-NMR (CF3COOD) δ/ ppm: 158.97 (lactam C=O), 149.78 (Cq), 148.15 (C-S), 139.60 (CHAr), 134.26 (Cq), 133.20 (CHAr), 132.92 (CHAr), 115.58 (CN), 32.47 (NCH3), 20.61 (SCH2).
3-[(4-Methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl)sulfanyl]propionic acid (4k).
(Alkylating agent: chloropropionic acid): yield 1.58 g (52%), M.p. 216-217 °C; Calculated for C13H12N4O3S (304.32): 51.31% C, 3.97% H, 18.41% N, 10.53% S, Found: 51.19% C, 3.83% H, 18.14% N, 10.49% S; FTIR, ṽ /cm-1: 1687 (C=O), 1711 (C=O acid), 3304-2908 (OH-acid broad), 2987, 2955 (CH), 1613 (C=N); 1H-NMR (DMSO-d6) δ/ ppm: 8.48-8.45 (1H, m, ArH), 8.25-8.22 (1H, m, ArH), 7.96-7.91 (1H, m, ArH), 7.63-7.58 (1H, m, ArH), 3.58 (3H, s, NCH3), 3.42 (2H, t, SCH2, J= 6.72 Hz), 2.77 (2H, t, CH2CO, J= 6.72 Hz); 13C-NMR (DMSO-d6) δ/ ppm: 172.51 (acid C=O), 158.13 (lactam C=O), 149.68 (Cq), 142.69 (C-S), 134.69 (CHAr), 133.39 (Cq), 128.91 (CHAr), 126.68 (CHAr), 117.08 (Cq), 115.61 (CHAr), 33.59 (SCH2), 29.05 (NCH3), 28.85 (CH2CO).
1-(Isopropylsulfanyl)-4-methyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (4l).
(Alkylating agent: isopropyl iodide) yield 1.09 g (40%), M.p. 148-149 °C; Calculated for C13H14N4OS (274.34): 56.92% C, 5.14% H, 20.42% N, 11.69% S, Found: 56.77% C, 5.10% H, 20.35% N, 11.57% S; FTIR, ṽ /cm-1: 1684(C=O), 2987, 2961 (CH), 1614 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.66-8.63 (1H, m, ArH), 8.44-8.41 (1H, m, ArH), 7.83-7.78 (1H, m, ArH), 7.57-7.52 (1H, m, ArH), 4.13-3.99 (1H, m, SCH), 3.80 (3H, s, NCH3), 1.51 (6H, d, CH(CH3)2, J= 6.72 Hz); 13C-NMR (CDCl3) δ/ ppm: 158.86 (lactam C=O), 149.94 (Cq), 144.54 (C-S), 134.52 (CHAr), 134.11 (Cq), 130.01 (CHAr), 129.89 (CHAr), 126.89 (CHAr), 117.61 (Cq), 116.00 (CHAr), 40.80 (SCH), 29.79 (NCH3), 23. 48 (CH(CH3)2).
General Procedure for N-Alkylation with Alkyl Halides
To a mixture of triazolo derivative 3 (2.32 g, 0.01 mol) and sodium hydroxide (0.02 mol) in absolute ethyl alcohol the appropriate alkyl halide (0.01 mol) was added. The reaction mixture was heated under reflux for 6h, concentrated under reduced pressure. The solid obtained was filtered, and crystallized from ethyl alcohol.
Ethyl-3-(4-methyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-2-yl) propanoate (5a).
(Alkylating agent: ethyl-3-chloropropanoate): yield 1.43 g (45%); M.p. 120-121 °C; Calculated for C15H16N4O3S (332.38): 54.21% C, 4.85% H, 16.86% N, 9.65% S, Found: 54.15% C, 4.76% H, 16.82% N, 9.57% S; FTIR, ṽ /cm-1: 1686(C=O), 1725 (C=O ester), 3087, 2977 (CH), 1631 (C=N); 1H-NMR (CDCl3) δ/ ppm: 10.33-10.31 (1H, m, ArH), 8.36-8.34 (1H, m, ArH), 7.82-7.79 (1H, m,), 7.58-7.56 (1H, m, ArH), 4.59 (1H, t, NCH2, J= 7.1 Hz), 4.18 (1H, q, OCH2, J= 7.1 Hz), 3.61(3H, s, NCH3), 2.93 (2H, t, CH2CO, J= 7.1 Hz), 1.29 (3H, t, CH2CH3, J= 7.1 Hz); 13C-NMR (CDCl3) δ/ ppm: 170.51 (C=O), 161.72 (C=S), 158.36 (lactam C=O), 144.36 (Cq), 135.13 (Cq), 134.45 (CHAr), 129.00 (CHAr), 127.25 (CHAr), 116.92 (Cq), 116.55 (CHAr), 60.96 (OCH2), 44.32 (NCH2), 32.23 (CH2CO), 29.02 (NCH3), 14.17 (CH2CH3).
3-(4-Methyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-2yl)propionitrile (5b).
(Alkylating agent: 3-chloropropanenitrile): yield 1.20 g (40%), M.p. 230-231 °C; Calculated for C13H11N5OS (285.32): 54.73% C, 3.89% H, 24.55% N, 11.24% S, Found: 54.69% C, 3.71% H, 24.45% N, 11.12% S; FTIR, ṽ /cm-1: 1687 (C=O), 2248 (CN), 3005, 2978, 2945 (CH), 1632 (C=N); 1H-NMR (CDCl3) δ/ ppm: 10.27-10.26 (1H, m, ArH), 8.39-8.37 (1H, m, ArH), 7.85-7.81 (1H, m, ArH), 7.60-7.56 (1H, m, ArH), 4.59 (2H, t, NCH2, J= 6.85 Hz), 3.64 (3H, s, NCH3), 3.03 (2H, t, CH2CN, J= 6.85 Hz); 13C-NMR (CDCl3) δ/ ppm: 162.20 (C=S), 158.36 (lactam C=O), 144.76 (Cq), 135.13 (Cq), 134.56 (CHAr), 129.19 (CHAr), 127.55 (CHAr), 116.96 (Cq), 116.32 (CHAr), 115.23 (CN), 44.14 (NCH2), 29.10 (NCH3), 16.09 (CH2CN); Mass spectrum, m/z (Ir/%):286 (18), 285 (100), 245 (34), 232 (29), 199 (3), 174 (5), 162 (12), 144 (8).
Aryl substitution reaction
1-[(2,4-Dinitrophenyl)sulfanyl]-4-methyl-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (6).
1-Chloro-2,4-dinitrobenzene (0.01 mol) was added to an alcoholic solution of triazolo derivative 3 (2.32 g, 0.01 mol) and triethylamine (2 mL, 0.02 mol) in ethyl alcohol. The reaction mixture was heated under reflux for 15 min to immediately give the product. The solid obtained was filtered, and crystallized from ethyl alcohol to give 3.26 g (82%) of the title compound; M.p. 319-320 °C; Calculated for C16H10N6O5S (398.35): 48.24% C, 2.53% H, 21.10% N, 8.05% S, Found: 48.21% C, 2.49% H, 20.88% N, 7.91% S; FTIR, ṽ /cm-1: 1693(C=O), 3074, 2955 (CH), 1614 (C=N); 1H-NMR (CF3COOD) δ/ ppm: 9.56-9.54 (1H, m, ArH), 9.09-9.06 (1H, m, ArH), 8.91-8.89 (1H, m, ArH), 8.66-8.63 (1H, m, ArH), 8.27-8.22 (1H, m, ArH), 8.15-8.10 (1H, m, ArH), 7.70-7.67 (1H, m, ArH), 4.31 (3H, s, NCH3); 13C-NMR (CF3COOD) δ/ ppm: 162.20 (lactam C=O), 152.12 (Cq), 149.74 (Cq), 148.16 (Cq), 143.79 (C-S), 139.67 (Cq), 139.37 (CHAr), 135.00 (Cq), 133.12 (CHAr), 132.86 (CHAr), 132.31 (CHAr), 131.22 (CHAr), 124.46 (CHAr), 119.60 (Cq), 118.39 (CHAr), 32.52 (NCH3).
Acylation reactions: General procedure
The appropriate acyl chloride (benzoyl chloride, pivaloyl chloride or ethyl chloroformate) (0.01 mol) was added to a mixture of triazolo derivative 3 (2.32 g, 0.01 mol) and triethylamine (2 mL, 0.02 mol) solution in benzene (30 mL). The reaction mixture was heated under reflux for 15 min. and then concentrated under reduced pressure. The solid obtained was filtered, and crystallized from ethyl alcohol. The obtained product was a mixture of both 7 and 8 and was not further purified. The purity of the isomers was determined from the corresponding 1H-NMR spectra. Compounds 7a and 7b were not isolated but detected only from the 1H-NMR spectra, FTIR spectroscopy and by the comparison with the 1H-NMR spectra of the pure 8a and 8b. The mixture of compounds obtained from the above general procedure 7a, 8a or 7b, 8b was heated under reflux in chloroform for 30 min. to afford only the pure acyl derivatives 8a or 8b.
Ethyl-[(4-methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl)sulfanyl]methanoate (7a).
FTIR, ṽ /cm-1: 1708 (C=O), 1743 (C=O ester), 3063, 2980 (CH), 1635 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.74-8.71 (1H, m, ArH), 8.45-8.42 (1H, m, ArH), 7.81-7.78 (1H, m, ArH), 7.56-7.53 (1H, m, ArH), 4.33 (2H, q, OCH2, J= 7.0 Hz), 3,82 (3H, s, NCH3), 1,29 (3H, t, CH2CH3, J= 7.0 Hz).
4-Methyl-5-oxo-4,5-dihydro[1,2,4]triazolo[4,3-a]quinazolin-1-yl-2,2-dimethylpropanethioate (7b).
FTIR, ṽ /cm-1: 1691(C=O), 1743 (C=O), 3002, 2978 (CH), 1645 (C=N); 1H-NMR (CDCl3) δ/ ppm: 8.62-8.59 (1H, m, ArH), 8.45-8.43 (1H, m, ArH), 7.83-7.79 (1H, m, ArH), 7.58-7.55 (1H, m, ArH), 3.84 (3H, s, NCH3), 1.61 (9H, s, C(CH3)3).
Ethyl-4-methyl-5-oxo-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-2-carboxylate (8a).
Yield 1.64 g (54%), M.p. 181-182°C; Calculated for C13H12N4O3S (304.32): 51.31% C, 3.97% H, 18.41% N, 10.53% S, Found: 51.21% C, 3.95% H, 18.33% N, 10.42% S; FTIR, ṽ /cm-1: 1689 (C=O), 1778 (ester C=O), 3063, 2980 (CH), 1635 (C=N); 1H-NMR (CDCl3) δ/ ppm: 10.41-10.38 (1H, m, ArH), 8.36-8.33 (1H, m, ArH), 7.81-7.78 (1H, m, ArH), 7.57-7.53 (1H, m, ArH), 4.55 (2H, q, OCH2, J= 7.0 Hz), 3.66 (3H, s, NCH3), 1.50 (3H, t, CH2CH3, J= 7.0 Hz); 13C-NMR (CDCl3) δ/ ppm: 176.08 (C=O), 164.75 (C=S), 158.48 (lactam C=O), 149.04 (Cq), 145.01 (Cq), 135.18 (Cq), 134.85 (CHAr), 129.47 (CHAr), 127.91 (CHAr), 117.17 (Cq), 116.55 (CHAr), 65.30 (OCH2), 29.36 (NCH3), 14.14.36 (CH2CH3).
2-(2,2-Dimethylpropanoyl)-4-methyl-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin- 5-one (8b).
Yield 1.42 g (45%), M.p. 197-198°C; Calculated for C15H16N4O2S (316.38): 56.95% C, 5.10% H, 17.71% N, 10.13% S, Found: 56.73% C, 4.97% H, 17.65% N, 9.96% S; FTIR, ṽ /cm-1: 1691(C=O), 1743 (C=O), 3002, 2978 (CH), 1645 (C=N); 1H-NMR (CDCl3) δ/ ppm: 10.50-10.47 (1H, m, ArH), 8.40-8.37 (1H, m, ArH), 7.84-7.79 (1H, m, ArH), 7.61-7.56 (1H, m, ArH), 3.67 (3H, s, NCH3), 1.52 (9H, s, C(CH3)3); 13C-NMR (CDCl3) δ/ ppm: 176.08 (C=O), 164.71 (C=S), 158.65 (lactam C=O), 143.89 (Cq), 135.20 (Cq), 134.79 (CHAr), 129.50 (CHAr), 127.88 (CHAr), 117.09 (Cq), 116.84 (CHAr), 29.87 (C(CH3)3), 29.24 (NCH3), 27.40 (C(CH3)3).
2-Benzoyl-4-methyl-1-thioxo-1,2,4,5-tetrahydro[1,2,4]triazolo[4,3-a]quinazolin-5-one (8c).
Yield 1.17 g (35%), M.p. 194-195°C; Calculated for C17H12N4O2S (336.37): 60.70% C, 3.60% H, 16.66% N, 9.53% S, Found: 60.59% C, 3.45% H, 16.57% N, 9.49% S; FTIR, ṽ /cm-1: 1693 (C=O), 1720 (C=O), 2987, 2955 (CH), 1640 (C=N); 1H-NMR (CDCl3) δ/ ppm: 10.36-10.33 (1H, m, ArH), 8.38-8.36 (1H, m, ArH), 7.99-7.93 (2H, m, ArH), 7.82-7.78 (1H, m, ArH), 7.66-7.46 (4H, m, ArH), 3.60 (3H, s, NCH3); 13C-NMR (CDCl3) δ/ ppm: 166.40 (C=O), 165.04 (C=S), 158.55 (lactam C=O), 142.41 (Cq), 135.08 (Cq), 134.76 (CHAr), 134.01 (CHAr), 131.98 (CHq), 131.14 (CHAr), 129.46 (CHAr), 128.57 (CHAr), 127.92 (CHAr), 117.09 (Cq), 116.66 (CHAr), 29.32 (NCH3).


{kind=link}
{kind=link}
{kind=link}
{kind=link}