Synthesis of new Bis- and Tetra-Acridines

UMR 6009, Laboratoire de Valorisation de la Chimie Fine, Université d'Aix-Marseille III, Av. Escadrille Normandie Niemen, Case 552, 13397 Marseille cedex 20, France
*
Author to whom correspondence should be addressed.
Molecules 2001, 6(8), 673-682; https://doi.org/10.3390/60800673
Submission received: 10 April 2001
/
Revised: 11 July 2001
/
Accepted: 11 July 2001
/
Published: 31 July 2001
Abstract
:A new series of bis- and tetra-acridines has been prepared from 4-(bromo-methyl)acridine; some of them exhibited encouraging in vitro cytotoxic activities against murine cell lines.
Introduction
Acridine derivatives are well known therapeutic agents whose mutagenic properties depend on their ability to interact with nucleic acids. One mechanism of this interaction is the intercalation between the acridine moiety and the base pairs of the DNA helix.[1] Moreover the pharmacological activity of these intercalating drugs derives from their ability to inhibit the synthesis of nucleic acids by blocking the action of DNA metabolizing proteins.[2] To develop more effective antagonists of DNA metabolism, many chemists have made molecules combining two or more intercalating ligands. For instance, Denny and co-workers prepared tetra-acridinic derivatives [3] and demonstrated that the higher order structure of DNA can be controlled by a complexation using ligands with multiple binding sites. The biological response of such compounds could be of interest because the higher order DNA structure plays an essential part in the regulation of gene expression by the cooperative binding proteins.
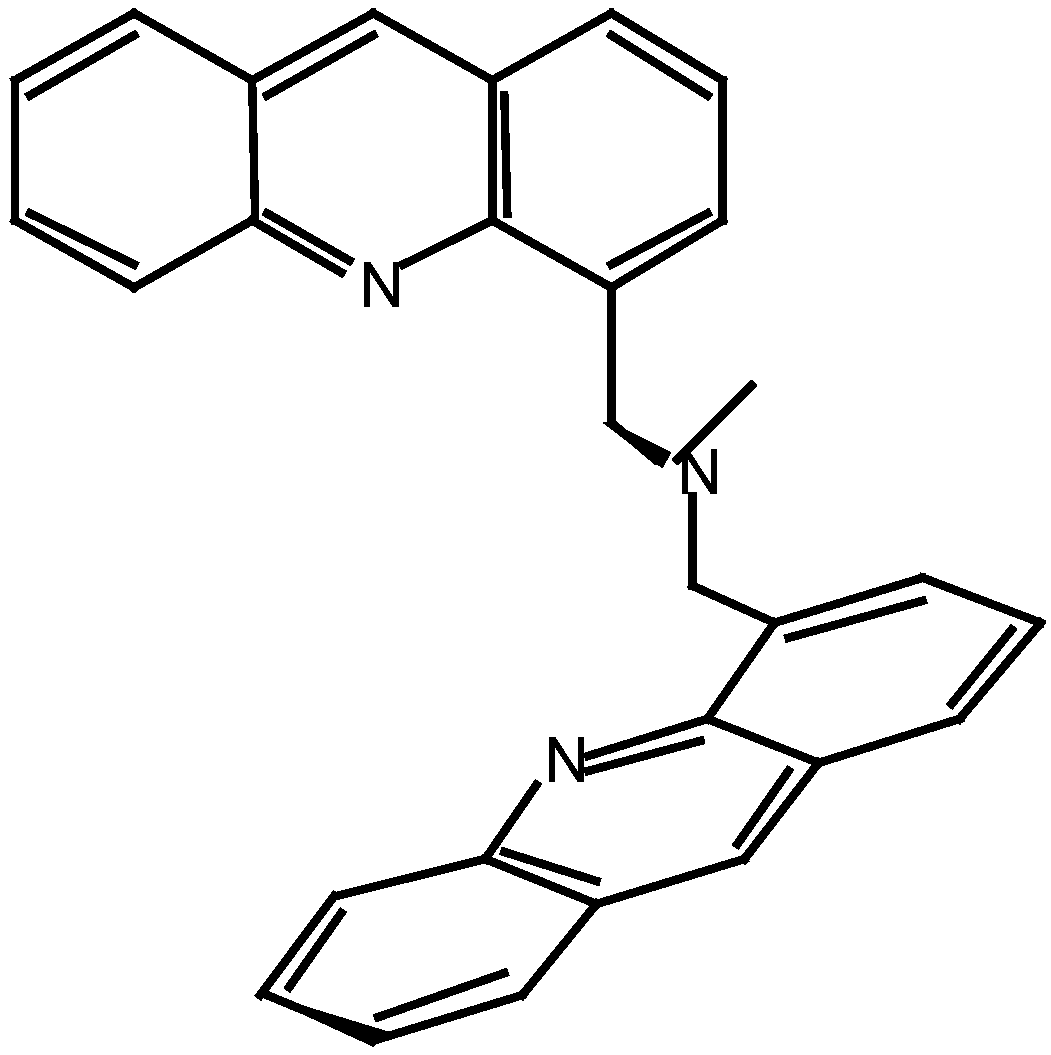
We prepared bis- and tetra-acridine compounds linked by a short nitrogen chain, with acridine moities closed enough to avoid the self-stacking of the aromatic planes; consequently all the synthesized compounds include a specific fragment like the N-bisacridine represented in Scheme 1.
Scheme 1.

Results and Discussion
Our synthetic pathway was based first on the preparation in 5 steps of the 4-(bromomethyl)acridine 5, followed by N-alkylation with different amines. First, the condensation of ortho-toluidine and 2-bromobenzoic acid by Ullmann condensation in ethylene glycol dimethyl ether with copper and anhydrous potassium carbonate gave the 2-[(2-methylphenyl)amino]benzoic acid (1) [4]. The cyclization of 1 in refluxing polyphosphoric acid (PPA) led finally to the 4-methyl-9(10H)-acridinone 2 by an intra-molecular Friedel-Crafts acylation [5]. The acridinone can be prepared alternatively with sulfuric acid or phosphorus oxychloride, but PPA is better because it induces less degradation of the anthranilic acid.
4-Methylacridine (4) was obtained in one pot after direct acidic oxidation of 4-methyl-acridane (3) (62%). Then, benzylic bromination of 4 was performed by a photochemical reaction. The use of N-bromosuccinimide (NBS) was described by Ledochowski [6] (50% yield), but we obtained better results (69% yield) with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 0.5 equiv.) [7] in cyclohexane and 6 hours reaction time (Scheme 2).
Scheme 2.

The dimeric and tetrameric polyacridine derivatives were obtained from the reaction of 4-(bromo-methyl)acridine (5) in dichloromethane or acetonitrile with different arylamines or aliphatic primary amines. Moreover, dimers and tetramers can be obtained selectively using a specific reactant ratio as described in Table 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amine equivalent | 0.5 equ. | 1 equ. | 1.5 equ. |
| primary amine | - | - | dimer |
| primary diamine | tetramer | dimer | - |
| secondary diamine | - | - | dimer |
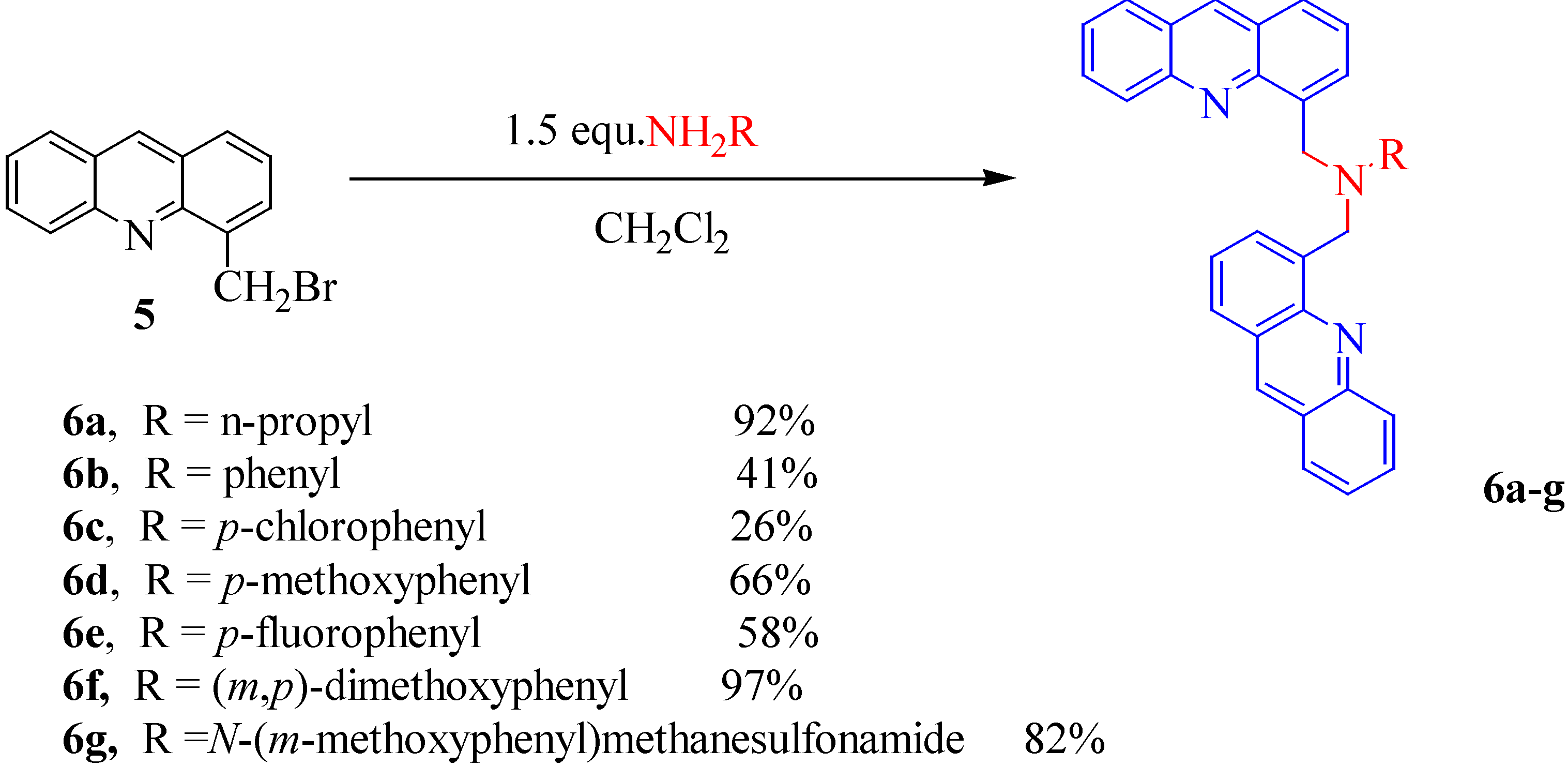
To obtain the dimeric acridine derivatives 6a-g we used aliphatic and aromatic primary amines (1.5 equivalent of amine) in dichloromethane; the yields are 41-97 % after purification, except for the N,N-bis[methyl(4'-acridinyl)]-p-chlorophenyl 6c preparation (26 %) (Scheme 3).
Scheme 3.

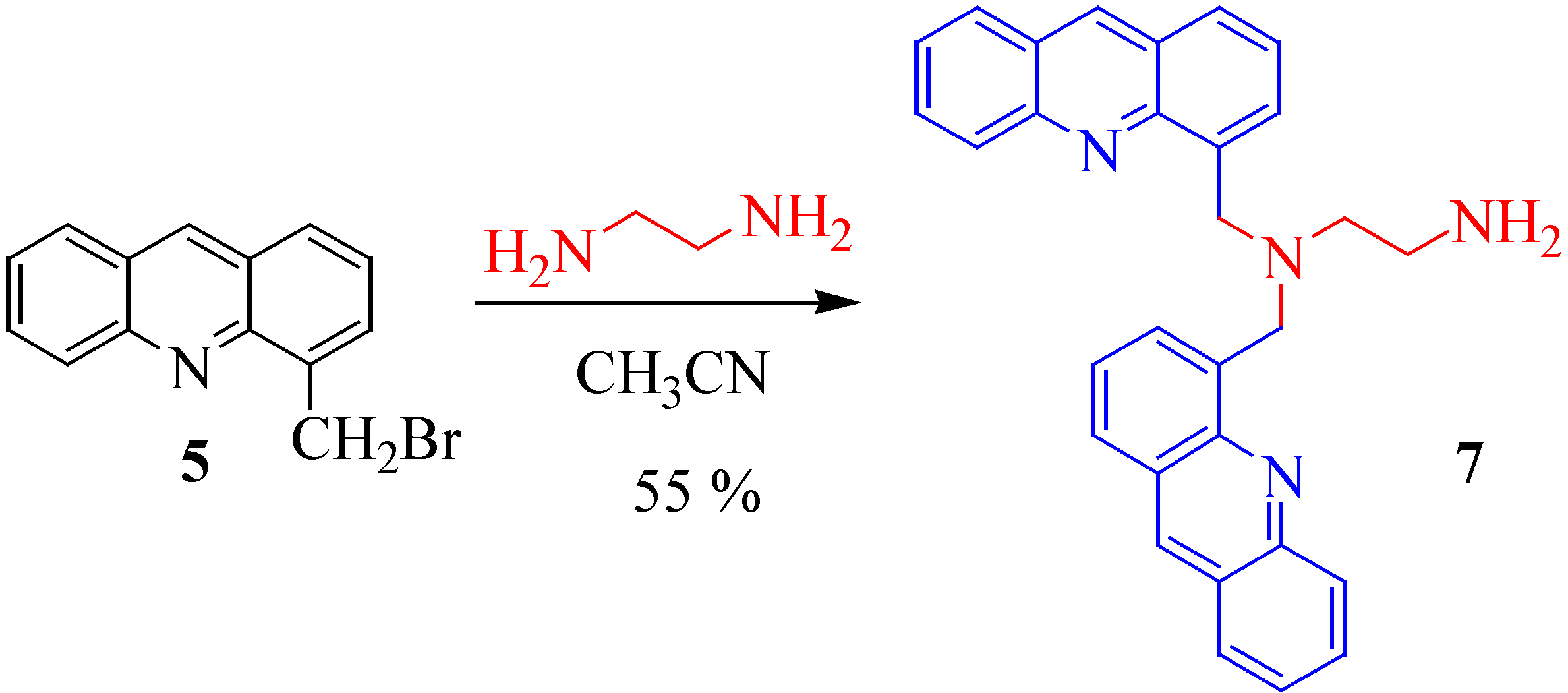
Using only one equivalent of the ethylene diamine with 5 in acetonitrile we obtained only one polycycle, the N,N-bis[methyl(4'-acridinyl)]-aminoethylamine 7 in 55 % yield (Scheme 4).
Scheme 4.

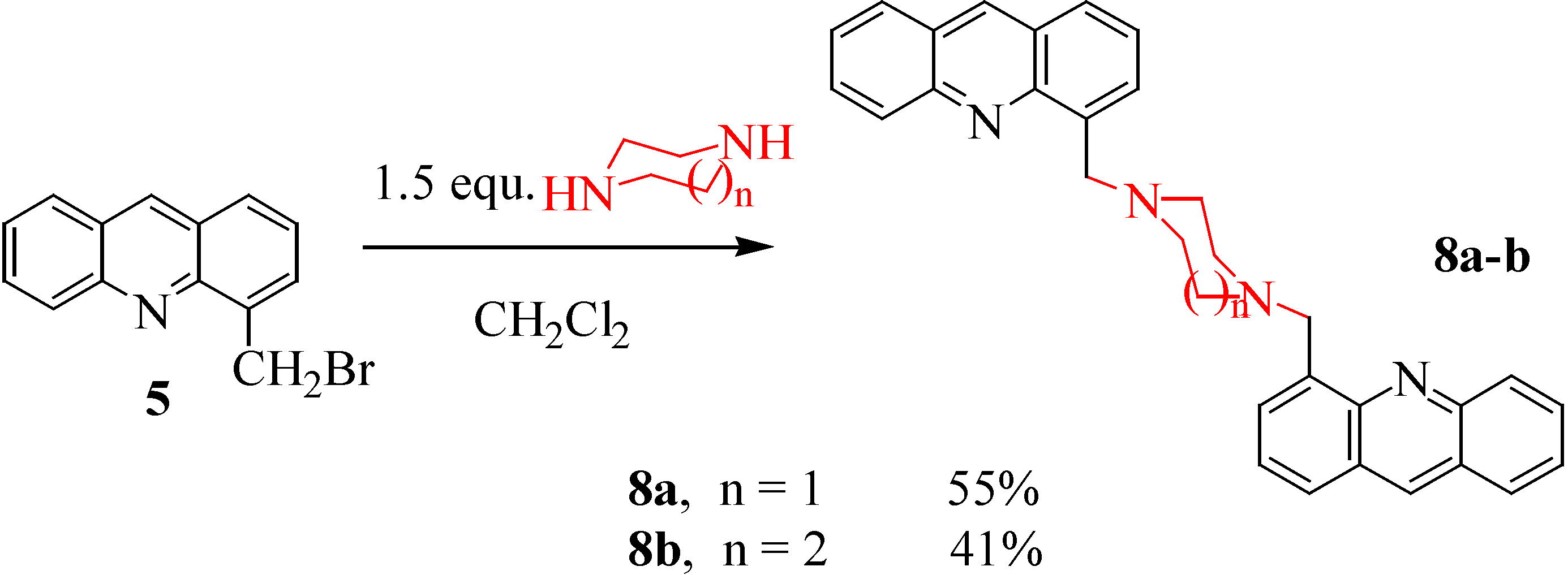
With two cyclic diamines, such as piperazine and homopiperazine, we obtained two dimers 8a-b, in yields of 55 and 41% respectively (Scheme 5).
Scheme 5.

Tetrameric N,N,N',N'-tetra[methyl(4'-acridinyl)]-diaminoalcanes 9a-e were obtained using different diamines. With only 0.5 equivalent of amine, only tetrameric acridines were recovered, in 52 – 89% yields (Scheme 6).
Scheme 6.

Biological Activity
The cytotoxicities of ten compounds (6a, 6d-g, 7, 9a, 9c-e) were determined in a panel of cell lines. The murine L1210 leukemia and A549 cell lines were used as a straightforward comparison of antiproliferative properties. Dimers 6e and 6f and tetramer 9a had the best results against DNA reparation, while polyacridine 9c had the least action against the L1210 leukemia cell line.
Conclusions
We propose an easy way to prepare a new class of polyacridinic derivatives using 4-(bromo-methyl)acridines. Most of them display some biological activity and we are now expanding the scope of this approach to the synthesis of other polyacridines.
Experimental
General
The NMR spectra were recorded on a Bruker AM 200 spectrometer working at 200.13 MHz; Homonuclear (COSY) and heteronuclear (HMBC, HMQC) bidimensional spectra were performed on a Bruker AMX 400. The chemical shifts are reported in δ values for CDCl3 or DMSO-d6 solutions. Melting points were measured on an Electrothermal apparatus and are uncorrected. Reagents and solvents were purchased from commercial suppliers.
2-[(2-Methylphenyl)amino]benzoic acid (1). Compound 1 was obtained from the condensation of 2-bromobenzoic acid (15 g, 74 mmol) and o-toluidine (11 g, 0.102 mol), in the presence of copper catalyst (0.26 g, 4.09 mmol) and anhydrous potassium carbonate (13.95 g, 0.1 mol), all in ethylene glycol dimethyl ether (80 mL). After work-up a white powder was obtained (13.5 g, 67%); mp 185°C; lit. [8] mp 185°C; 1H-NMR (DMSO-d6) δ: 2.19 (s, 3H, CH3), 6.72 (dd, 1H, J = 8.0, 7.2 Hz), 6.85 (dd, 1H, J = 8.1, 1.3 Hz), 7.05 (ddd, 1H, J = 8.2, 7.4, 1.4 Hz), 7.28 (m, 4H), 7.88 (dd, 1H, J = 8.0, 1.3 Hz), 9.49 (s, 1H), 13.00 (s, 1H); 13C-NMR (DMSO-d6) δ: 18.04, 109.71, 113.74, 116.52, 124.89, 125.20, 126.73, 131.13, 132.53, 133.49, 135.78, 138.54, 149.74, 174.23; Anal. Calcd for C14H13NO2: C, 73.99; H, 5.77; N, 6.16; Found: C, 74.25; H, 5.50; N, 6.49.
4-Methyl-9(10H)]acridinone (2). A mixture of 1 (4 g, 17 mmol) and PPA (50 g) was refluxed for 3 hours at 120°C. Next the solution was poured into cold water (400 mL). After treatment with an ammonia solution (36%) and filtration of the suspension, the precipitate was dried and refluxed for 1 hour in DMF (150 mL). After filtration, the acridinone was precipitated in cold water (400 mL), and filtered to yield a yellow powder (4.41 g, 94%); mp 346°C; lit. [9] mp 346°C; 1H-NMR (DMSO-d6) δ: 2.59 (s, 3H, CH3), 7.16 (dd, 1H, J = 8.1, 7.5 Hz), 7.28 (ddd, 1H, J = 8.0, 7.2, 1.3 Hz), 7.58 (dd, 1H, J = 7.5, 1.2 Hz), 7.71 (ddd, 1H, J = 8.1, 7.4, 1.1 Hz), 7.92 (dd, 1H, J = 8.1, 1.2 Hz), 8.12 (dd, 1H, J = 8.0, 1.3 Hz), 8.21 (dd, 1H, J = 8.1, 1.4 Hz), 10.6 (s, 1H); 13C-NMR (DMSO-d6) δ: 17.82, 118.14, 120.33, 120.69, 120.69, 121.24, 123.93, 125.28, 125.73, 133.20, 134.08, 139.51, 141.07, 177.02; Anal. Calcd for C14H11NO: C, 80.36; H, 5.30; N, 6.69; Found: C, 74.25; H, 5.50; N, 6.49.
4-Methylacridine (4). A mixture of 2 (3g, 14 mmol) in butan-1-ol (77 mL) was stirred at 110°C and a solution of sodium (6g, 0.26 mol) dissolved in butan-1-ol (10 mL) was added dropwise. The mixture was then concentrated and the residual solid was recrystallized from water to yield a pale yellow powder of acridane 3. Compound 3 was stirred with nitric acid (69%, 83 mL) for 2 hours at room temperature. The mixture was diluted with water (120 mL) and stirred for an additional 1 hour. After filtration, the filtrate was made basic with ammonia to pH 8 and filtered. The precipitate was dried, dissolved in hot ethanol (60 mL), and filtered. The organic phase was poured into water (200 mL), acidified with HCl (10 mL), and filtered. Next the solution was made basic with an ammonia solution (16%) and filtered to yield a yellow powder (1.70 g, 62%); mp 89°C; lit. [10] mp 89°C; 1H-NMR (CDCl3) δ: 2.96 (s, 3H, CH3), 7.43 (dd, 1H, J = 8.0, 7.5 Hz), 7.53 (ddbr, 1H, J = 8.1, 7.3 Hz), 7.63 (dd, 1H, J = 7.5, 1.4 Hz), 7.73 (ddd, 1H, J = 8.1, 7.2, 1.5 Hz), 7.86 (dd, 1H, J = 8.1, 1.4 Hz), 7.99 (dbr, 1H, J = 8.1 Hz), 8.29 (dd, 1H, J = 8.1, 7.1 Hz), 8.73 (s, 1H); 13C-NMR (CDCl3) δ: 18.40, 125.41, 125.41, 126.14, 126.28, 126.46, 127.87, 129.42, 129.62, 129.87, 135.79, 137.11, 148.29, 148.53; Anal. Calcd for C14H11N: C, 87.01; H, 5.74; N, 7.25; Found: C, 87.19; H, 5.61; N, 7.33.
4-Bromomethylacridine (5). A mixture of 4 (3.15 g, 16 mmol) and DBDMH, (1,3-dibromo-5,5-dimethylhydantoin), (2.5 g, 8.74 mmol) in cyclohexane (160 mL) was placed in a three-necked flask. The solution was then warmed with a 500-Watt tungsten lamp under a nitrogen atmosphere and stirred for 6 hours. After filtration, the solution was cooled and the obtained yellow needles were filtered to give 5 (3.1 g, 69%); mp 165°C; lit. [11] mp 165°C; 1H-NMR (CDCl3) δ : 5.41 (s, 2H, CH2), 7.45 (dd, 1H, J = 8.0, 7.2 Hz), 7.53 (ddbr, 1H, J = 8.1, 7.4 Hz), 7.78 (ddd, 1H, J = 8.2, 7.3, 1.5 Hz), 7.90 (dd, 1H, J = 7.2, 1.3 Hz), 7.94 (dd, 1H, J = 7.2, 1.3 Hz), 7.96 (dbr, 1H, J = 8.0 Hz), 8.29 (dbr, 1H, J = 8.2 Hz), 8.70 (s, 1H); 13C-NMR (CDCl3) δ: 30.21, 125.37, 126.03, 126.10, 126.79, 128.03, 129.26, 130.23, 130.35, 131.14, 136.15, 136.15, 146.48, 148.75; Anal. Calcd for C14H10NBr: C, 61.79; H, 3.70; N, 5.15; Found: C, 62.03; H, 3.58; N, 5.41.
N,N'-Bis[methyl(4'-acridinyl)]propylamine (6a). A mixture of 5 (0.2 g, 0.73 mmol) and propylamine (0.065 g, 1.1 mmol) in dichloromethane (8 mL) was stirred under reflux for 3 hours. The solvent was evaporated. The resulting oil was recovered and dissolved in dilute HCl (2%, 5 mL) to yield 6a as a brown powder after filtration (0.15 g, 92%); mp 124°C; 1H-NMR (CDCl3) δ: 1.23 (t, 3H, CH3, J = 6.9 Hz), 2.29 (m, 2H, CH2), 3.91 (t, 2H, CH2, J = 6.9 Hz), 5.39 (d, 2H, CH2, J = 14.1 Hz), 5.69 (d, 2H, CH2, J = 14.1 Hz), 7.21 (m, 2H), 7.22 (m, 2H), 7.24 (t, 2H, J = 7.7 Hz), 7.34 (m, 2H), 7.60 (dbr, 2H, J = 8.0 Hz), 7.67 (dbr, 2H, J = 8.1 Hz), 7.97 (dbr, 2H, J = 8.0 Hz), 8.30 (s, 2H); 13C-NMR (CDCl3) δ: 11.46, 19.11, 57.02, 58.58, 125.28, 125.28, 126.00, 126.08, 127.47, 127.91, 128.03, 129.61, 130.58, 134.45, 136.70, 146.11, 147.12; Anal. Calcd for C31H27N3: C, 84.32; H, 6.16; N, 9.52; Found: C, 83.97; H, 6.48; N, 9.44.
N,N'-Bis[methyl(4'-acridinyl)]aniline (6b). A mixture of 5 (0.2 g, 0.73 mmol) and aniline (0.105 g, 1.1 mmol) in dichloromethane (8 mL) was refluxed and stirred for 3 hours. The mixture was filtered and the solvent evaporated. The resulting oil was dissolved in dilute HCl (2%, 5 mL) to yield a brown powder of 6b (0.073 g, 41%); mp 139°C; 1H-NMR (DMSO-d6) δ: 5.60 (s, 4H, CH2), 7.08 (d, 2H, J = 7.8 Hz), 7.58 (t, 1H, J = 7.8 Hz), 7.60 (dd, 2H, J = 8.8, 7.4 Hz), 7.73 (m, 4H), 7.76 (dbr, 2H, J = 8.0 Hz), 7.78 (ddd, 2H, J = 8.2, 7.1, 1.2 Hz), 8.13 (dbr, 2H, J = 8.1 Hz), 8.18 (dbr, 4H, J = 10.2 Hz), 9.16 (s, 2H); 13C-NMR (DMSO-d6) δ: 51.78, 111.93, 116.04, 125.77, 126.09, 126.16, 126.46, 127.37, 127.58, 128.62, 128.95, 129.26, 130.90, 134.83, 137.13, 146.81, 147.40, 148.32; Anal. Calcd for C34H25N3: C, 85.87; H, 5.30; N, 8.84; Found: C, 85.99; H, 5.03; N, 8.76.
N,N'-Bis[methyl(4'-acridinyl)]-p-chloroaniline (6c). Prepared as described for 6a but with 5 (0.2 g, 0.73 mmol) and p-chlorophenylamine (0.140 g, 1.1 mmol). Work up yielded a brown powder which was dissolved in dichloromethane and precipitated with diethyl ether to afford the title compound (0.048 g, 26%); mp 120°C; 1H-NMR (DMSO-d6) δ: 5.59 (s, 4H, CH2), 6.69 (d, 2H, J = 8.6 Hz), 7.08 (d, 2H, J = 8.5 Hz), 7.65 (m, 6H), 7.84 (ddd, 2H, J = 8.1, 7.2, 1.4 Hz), 8.09 (d, 2H, J = 8.2 Hz), 8.17 (d, 4H, J = 8.1 Hz), 9.14 (s, 2H); 13C-NMR (DMSO-d6) δ: 51.68, 113.23, 115.29, 119.26, 125.61, 125.96, 126.08, 126.30, 126.99, 128.48, 128.72, 129.03, 130.67, 134.60, 136.76, 146.85, 147.21, 147.47; Anal. Calcd for C34H24N3Cl: C, 80.07; H, 4.74; N, 8.24; Found: C, 80.34; H, 4.41; N, 8.01.
N,N'-Bis[methyl(4'-acridinyl)]-p-anisidine (6d). As described for 6a but with 5 (0.2 g, 0.73 mmol) and p-anisidine (0.140 g, 1.1 mmol). After work up a brown powder of 6d was recovered (0.122 g, 66%); mp 140°C; 1H-NMR (DMSO-d6) δ: 3.68 (s, 3H, OCH3), 5.60 (s, 4H, CH2), 6.95 (d, 2H, J = 8.8 Hz), 7.31 (d, 2H, J = 8.1 Hz), 7.55 (m, 8H), 7.80 (d, 2H, J = 8.2 Hz), 8.08 (m, 4H), 9.08 (s, 2H); 13C-NMR (DMSO-d6) δ: 55.55, 60.24, 115.37, 122.67, 125.34, 125.51, 126.21, 126.25, 126.91, 127.78, 128.12, 130.02, 130.91, 134.16, 134.23, 137.29, 145.96, 147.19, 160.10; Anal. Calcd for C35H27N3O: C, 83.14; H, 5.38; N, 8.31; Found: C, 83.33; H, 5.19; N, 8.24.
N,N'-Bis[methyl(4'-acridinyl)]-p-fluoroaniline (6e). A mixture of 5 (0.2 g, 0.73 mmol) and p-fluoro-phenylaniline (0.122 g, 1.1 mmol) in dichloromethane (8 mL) was stirred under reflux for 3 hours. The solvent was evaporated, and the resulting oil was dissolved in dichloromethane and washed with dilute HCl (2%, 10 mL). The oil recovered after evaporation was dissolved in dilute HCl (2%, 5 mL) to yield a brown powder of 6e (0.105 g, 58%); mp 174°C; 1H-NMR (CDCl3) δ: 5.69 (s, 4H, CH2), 6.85 (dd, 4H, J = 11.0, 8.1 Hz), 7.49 (ddd, 4H, J = 8.1, 7.2, 1.4 Hz), 7.69 (dd, 2H, J = 8.0, 7.3 Hz), 7.91 (m, 6H), 8.23 (d, 2H, J = 8.1 Hz), 8.73 (s, 2H); 13C-NMR (CDCl3) δ: 54.14, 113.80, 116.18, 116.62, 126.32, 126.41, 127.00, 127.17, 128.09, 128.75, 129.12, 130.02, 130.85, 134.70, 136.96, 147.98, 148.29, 148.59; Anal. Calcd for C34H24FN3: C, 82.74; H, 4.90; N, 8.51; Found: C, 82.98; H, 4.96; N, 3.40.
N,N'-Bis[methyl(4'-acridinyl)]-(m,p)-dimethoxyaniline (6f). As described for 6a but with 5 (0.2 g, 0.73 mmol) and (m,p)-dimethoxyaniline (0.168 g, 1.1 mmol). A brown powder was recovered (0.192 g, 97%); mp 163°C; 1H-NMR (DMSO-d6) δ: 3.67 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 5.66 (s, 4H, CH2), 6.89 (s 1H), 7.54 (m, 10H), 7.83 (dd, 2H, J = 7.4, 1.4 Hz), 8.06 (dd, 2H, J = 8.1, 1.5 Hz), 8.10 (d, 2H, J = 8.3 Hz), 9.08 (s, 2H); 13C-NMR (DMSO-d6) δ: 56.07, 56.23, 58.59, 103.65, 111.08, 112.52, 125.61, 125.84, 126.10, 126.25, 126.43, 127.84, 128.74, 129.24, 129.84, 131.43, 132.64, 138.34, 146.01, 146.93, 150.06; Anal. Calcd for C36H29N3O2: C, 80.72; H, 5.46; N, 7.84; Found: C, 81.08; H, 5.35; N, 7.78.
N,N'-Bis[methyl(4'-acridinyl)]-N-(4-amino-3-methoxyphenyl)methanesulfonamide (6g). Prepared as described for 6a but using 5 (0.2 g, 0.73 mmol) and N-(4-amino-3-methoxyphenyl)methane-sulfonamide (0.238 g, 1.1 mmol). Work up yielded 6g as a brown powder (0.18 g, 82%); mp 161°C; 1H-NMR (DMSO-d6/TFA) δ: 2.99 (s, 3H, CH3), 3.02 (s, 3H, OCH3), 5.57 (s, 4H, CH2), 6.74 (d, 1H, J = 2.1 Hz), 7.01 (dd, 1H, J = 8.2, 2.0 Hz), 7.05 (d, 2H, J = 8.2 Hz), 7.46 (m, 6H), 7.77 (d, 2H, J = 8.1 Hz), 8.05 (dd, 2H, J = 7.3, 1.4 Hz), 8.09.(dd, 2H, J = 8.1, 1.2 Hz), 8.18 (d, 1H, J = 8.0 Hz), 9.03 (s, 2H); 13C-NMR (DMSO-d6/TFA) δ: 40.00, 55.88, 60.32, 102.84, 111.71, 124.28, 124.88, 125.47, 125.73, 126.53, 127.54, 127.89, 128.90, 130.85, 131.24, 134.31, 138.13, 141.18, 141.30, 146.24, 147.15, 152.42; Anal. Calcd for C36H30N4O3S: C, 72.22; H, 5.05; N, 9.36; Found: C, 72.07; H, 5.16; N, 9.43.
N,N'-Bis[methyl(4'-acridinyl)]aminoethylamine (7). A mixture of 5 (1 g, 3.67 mmol) and ethylene diamine (0.22 g, 3.67 mmol) in acetonitrile (40 mL) was stirred and refluxed for 2 hours. After cooling the solution was filtered. The precipitate was washed with water (60 mL) and a beige powder of 7 was thus obtained (0.45 g, 55%); mp 182°C; 1H-NMR (CDCl3) δ: 2.95 (t, 2H, CH2, J = 6.9 Hz), 3.17 (t, 2H, CH2, J = 6.9 Hz), 4.62 (s, 4H, CH2), 7.34 (dd, 2H, J = 8.1, 7.3 Hz), 7.43 (dd, 2H, J = 8.2, 7.3 Hz), 7.69 (dd, 2H, J = 8.1, 7.3 Hz), 7.77 (d, 2H, J = 8.1 Hz), 7.91 (dd, 2H, J = 7.4, 1.3 Hz), 7.92 (dd, 2H, J = 8.1, 1.3 Hz), 8.17 (d, 2H, J = 8.0 Hz), 8.65 (s, 2H); 13C-NMR (CDCl3) δ: 39.77, 54.62, 56.63, 125.46, 125.66, 126.36, 126.62, 127.28, 128.04, 129.68, 130.13, 130.19, 136.18, 137.31, 147.91, 148.32; Anal. Calcd for C30H26N4: C, 81.42; H, 5.92; N, 12.66; Found: C, 81.51; H, 6.04; N, 12.61.
N,N'-Bis[methyl(4'-acridinyl)]piperazine (8a). A mixture of 5 (0.18 g, 0.66 mmol) and anhydrous piperazine (0.06 g, 0.68 mmol) in acetonitrile (30 mL) was stirred and refluxed for 3 hours. The solvent was evaporated, and the resulting oil was diluted in water (5 mL) to yield compound 8a as a brown powder (0.085 g, 55%); mp 228°C; 1H-NMR (CDCl3) δ: 2.82 (s, 8H, CH2), 4.49 (s, 4H, CH2), 7.50 (dd, 2H, J = 8.1, 7.2 Hz), 7.51 (dd, 2H, J = 7.3, 1.2 Hz), 7.74 (ddd, 2H, J = 8.3, 7.0, 1.1 Hz), 7.89 (m, 4H), 7.96 (d, 2H, J = 8.1 Hz), 8.23 (d, 2H), 8.72 (s, 2H); 13C-NMR (CDCl3) δ: 52.81, 56.13, 124.51, 124.51, 125.30, 125.55, 125.88, 126.94, 128.22, 128.75, 128.99, 134.85, 135.36, 147.10, 147.30; Anal. Calcd for C32H28N4: C, 82.02; H, 6.02; N, 11.96; Found: C, 82.14; H, 6.18; N, 12.10.
N,N'-Bis[methyl(4'-acridinyl)]homopiperazine (8b). Prepared as described for 8a but with 5 (0.2 g, 0.73 mmol) and homopiperazine (0.075 g, 0.75 mmol). Work up yielded 8b as a brown powder (0.072 g, 41%); mp 170°C; 1H-NMR (CDCl3) δ: 2.64 (m, 2H, CH2), 3.83 (t, 4H, CH2, J = 6.8 Hz), 4.22 (s, 4H, CH2), 5.22 (s, 4H, CH2), 7.57 (m, 4H), 7.82 (ddd, 2H, J = 8.1, 7.2, 1.3 Hz), 7.98 (dd, 2H, J = 7.3, 1.5 Hz), 8.06 (dd, 2H, J = 8.2, 1.5 Hz), 8.40 (m, 4H), 8.80 (s, 2H); 13C-NMR (CDCl3) δ: 20.23, 47.64, 53.73, 55.51, 125.28, 126.01, 126.40, 126.40, 127.31, 128.52, 129.20, 131.01, 131.20, 134.73, 137.54, 146.60, 147.89; Anal. Calcd for C33H30N4: C, 82.13; H, 6.26; N, 11.61; Found: C, 82.20; H, 6.17; N, 11.73.
N,N,N',N'-Tetra[methyl(4'-acridinyl)]-1,2-diaminoethane (9a). A mixture of 5 (1 g, 3.67 mmol) and 1,2-diaminoethane (0.11 g, 1.8 mmol) in dichloromethane (40 mL) was stirred and refluxed for 3 hours. After cooling at room temperature, the solution was filtered. The precipitate was washed with water (60 mL) to afford 9a as a beige powder (0.45 g, 60%); mp 191°C; 1H-NMR (DMSO-d6) δ: 4.89 (s, 4H, CH2), 5.42 (s, 8H, CH2), 7.21 (dd, 4H, J = 7.5, 1.5 Hz), 7.31 (dd, 4H, J = 8.1, 7.3 Hz), 7.38 (dd, 4H, J = 8.2, 7.4 Hz), 7.48 (dd, 4H, J = 8.5, 7.4 Hz), 7.85 (m, 12H), 8.77 (s, 4H); 13C-NMR (DMSO-d6) δ: 51.48, 57.92, 124.97, 125.23, 125.78, 126.05, 127.66, 127.67, 128.31, 129.96, 130.66, 133.56, 137.42, 145.82, 146.65; Anal. Calcd for C58H44N6: C, 84.44; H, 5.37; N, 10.19; Found: C, 84.67; H, 5.48; N, 9.85.
N,N,N',N'-Tetra[methyl(4'-acridinyl)]-1,3-diaminopropane (9b). Prepared as described for 9a from 5 (0.2 g, 0.73 mmol) and 1,3-diaminopropane (0.027 g, 0.36 mmol). After filtration, the filtrate was evaporated. The resulting oil recovered was dissolved in water and the solution acidified (2 % HCl, 5 mL). A yellow powder of 9b was obtained after filtration (0.101 g, 66%); mp 129°C; 1H-NMR (DMSO-d6) δ: 3.45 (s, 2H, CH2), 4.33 (s, 4H, CH2), 5.36 (s, 8H, CH2), 7.02 (dd, 4H, J = 8.1, 7.4 Hz), 7.14 (dbr, 4H, J = 8.1 Hz), 7.25 (ddbr, 4H, J = 8.2, 7.2 Hz), 7.35 (ddbr, 4H, J = 8.2, 7.5 Hz), 7.70 (dd, 4H, J = 7.1, 1.4 Hz), 7.80 (d, 4H, J = 8.0 Hz), 7.82 (dd, 4H, J = 8.1, 1.4 Hz), 8.61 (s, 4H); 13C-NMR (DMSO-d6) δ: 19.57, 52.93, 56.88, 124.93, 125.14, 125.84, 126.03, 127.02, 127.42, 128.31, 130.00, 130.55, 133.76, 137.51, 145.64, 146.57; Anal. Calcd for C59H46N6: C, 84.46; H, 5.52; N, 10.02; Found: C, 84.66; H, 5.52; N, 9.82.
N,N,N',N'-Tetra[methyl(4'-acridinyl)]-1,4-diaminobutane (9c). Prepared as described for 9a but with 5 (0.2 g, 0.73 mmol) and 1,4-diaminobutane (0.032 g, 0.36 mmol). After filtration, the filtrate was evaporated. The resulting oil was dissolved in dilute HCl (2%, 5 mL) to yield 9c as a brown powder (0.115 g, 73%); mp 129°C; 1H-NMR (DMSO-d6) δ: 2.71 (sbr, 4H), 4.09 (sbr, 4H), 5.14 (sbr, 4H, CH2), 5.40 (sbr, 4H, CH2), 7.26 (ddbr, 4H, J = 8.2, 7.3 Hz), 7.36 (dbr, 4H, J = 8.1 Hz), 7.38 (dd, 4H, J = 8.1, 7.2 Hz), 7.40 (ddbr, 4H, J = 8.2, 7.2 Hz), 7.77 (dd, 4H, J = 7.3, 1.5 Hz), 7.81 (d, 4H, J = 8.0 Hz), 7.85 (dd, 4H, J = 8.1, 1.5 Hz), 8.70 (s, 4H); 13C-NMR (DMSO-d6) δ: 22.25, 56.06, 57.17, 124.99, 125.09, 125.76, 125.98, 127.13, 127.52, 128.20, 130.01, 130.73, 133.68, 137.44, 145.50, 146.49; Anal. Calcd for C60H48N6: C, 84.48; H, 5.67; N, 9.85; Found: C, 84.35; H, 5.77; N, 9.69.
N,N,N',N'-Tetra[methyl(4'-acridinyl)]-1,6-diaminohexane (9d). Prepared as described for 9a but with 5 (0.2 g, 0.73 mmol) and 1,6-diaminohexane (0.042 g, 0.36 mmol). After filtration, the filtrate was evaporated. The resulting oil was dissolved in dilute HCl (2%, 5 mL) to yield a brown powder of 9d (0.145 g, 89%); mp 145°C; 1H-NMR (DMSO-d6) δ: 1.84 (s, 4H, CH2), 2.38 (sbr, 4H, CH2), 3.72 (sbr, 4H, CH2), 5.03 (sbr, 4H, CH2), 5.37 (sbr, 4H, CH2), 7.25 (m, 8H), 7.35 (dd, 4H, J = 8.1, 7.5 Hz), 7.37 (ddbr, 4H, J = 8.0, 7.3 Hz), 7.70 (d, 4H, J = 8.1 Hz), 7.80 (d, 4H, J = 8.0 Hz), 7.82 (dd, 4H, J = 8.0, 1.4 Hz), 8.60 (s, 4H); 13C-NMR (DMSO-d6) δ: 24.34, 25.92, 56.69, 57.26, 124.98, 125.03, 125.76, 125.96, 127.30, 127.36, 128.29, 129.94, 130.68, 133.57, 137.34, 145.54, 146.51; Anal. Calcd for C62H52N6: C, 84.51; H, 5.95; N, 9.54; Found: C, 84.42; H, 6.01; N, 9.48.
N,N,N',N'-Tetra[methyl(4'-acridinyl)]-1,8-diaminooctane (9e). Prepared as described for 9a but with 5 (0.2 g, 0.73 mmol) and 1,8-diaminooctane (0.053 g, 0.36 mmol). After filtration, the filtrate was evaporated. The resulting oil was dissolved in dilute HCl (2%, 5 mL) to yield 9e as a brown powder (0.088 g, 52%); mp 136°C; 1H-NMR (DMSO-d6) δ: 1.42 (s, 4H), 1.66 (s, 4H), 2.25 (s, 4H), 3.82 (s, 4H), 4.91 (sbr, 4H, CH2), 5.28 (sbr, 4H, CH2), 7.24 (d, 4H, J = 8.3 Hz), 7.31 (dd, 4H, J = 8.1, 7.1 Hz), 7.33 (dd, 4H, J = 8.1, 7.5 Hz), 7.41 (ddbr, 4H, J = 8.1, 7.2 Hz), 7.62 (d, 4H, J = 7.5 Hz), 7.80 (d, 4H, J = 8.0 Hz), 7.82 (dd, 4H, J = 8.1, 1.4 Hz), 8.63 (s, 4H); 13C-NMR (DMSO-d6) δ: 24.57, 26.18, 28.65, 56.86, 57.37, 124.93, 125.02, 125.95, 127.34, 127.42, 127.79, 128.35, 129.91, 130.58, 133.39, 137.16, 145.62, 146.62; Anal. Calcd for C64H56N6: C, 84.55; H, 6.21; N, 9.24; Found: C, 84.63; H, 6.03; N, 9.36.
References
- Michaelis, L. Cold Spring Harbor Symp. Quant Biol. 1947, 12, 131. Heilweil, H.G.; Van Winkle, Q. J. Phys. Chem. 1955, 59, 939.
- Hayes, W. The Genetics of Bacteria and their Viruses; Blackwell Scientific Publications: Oxford (U.K.), 1968. [Google Scholar]
- Denny, W. A.; Atwell, G. J.; Baguley, B. C.; Wakelin, L.P.G. J. Med. Chem. 1985, 28, 1568.
- Ullmann, F. Chem. Ber. 1903, 36, 2384.
- Uhlig, F. Angew. Chem. 1954, 15, 435.
- Gruszecki, W.; Ledochowski, Z. Rocz. Chem. 1967, 41, 393.
- Reed, R. A. J. Chem. Soc. 1944, 679.
- Ullmann, F.; Bader, G. Justus Liebigs Ann. Chem. 1907, 355, 323.
- Uhlig, F. Angew. Chem. 1954, 15, 435.
- Reed, R. A. J. Chem. Soc. 1944, 679.
- Guszecki, W.; Borowski, E. Rocz. Chem. 1968, 42, 733.
- Sample availability: Not available
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
MDPI and ACS Style
Sourdon, V.; Mazoyer, S.; Pique, V.; Galy, J.-P. Synthesis of new Bis- and Tetra-Acridines. Molecules 2001, 6, 673-682. https://doi.org/10.3390/60800673
AMA Style
Sourdon V, Mazoyer S, Pique V, Galy J-P. Synthesis of new Bis- and Tetra-Acridines. Molecules. 2001; 6(8):673-682. https://doi.org/10.3390/60800673
Chicago/Turabian StyleSourdon, Valérie, Stéphane Mazoyer, Valérie Pique, and Jean-Pierre Galy. 2001. "Synthesis of new Bis- and Tetra-Acridines" Molecules 6, no. 8: 673-682. https://doi.org/10.3390/60800673