Experimental
General
All melting points are uncorrected. The IR spectra were recorded on a Nicolet FT-55X spectrophotometer. The 1H- NMR spectra were determined on a Varian FT-200 and Varian FT-300S instruments. All NMR spectra were obtained with the pulse sequence as partincluded with of the spectrometer's software and, unless specified otherwise, wasere determined in deuterochloroform solutions containing tetramethylsilane as the internal standard. with cChemical shifts (δ) are expressed in ppm downfield from tetramethylsilanethe reference peak. Mass spectra were recorded usingon a Jeol SX-102 mass spectrometer using the direct inlet system with an ionization energy of 70 eV, an emission current of 100 μA and and ion source temperature of 150 °C.
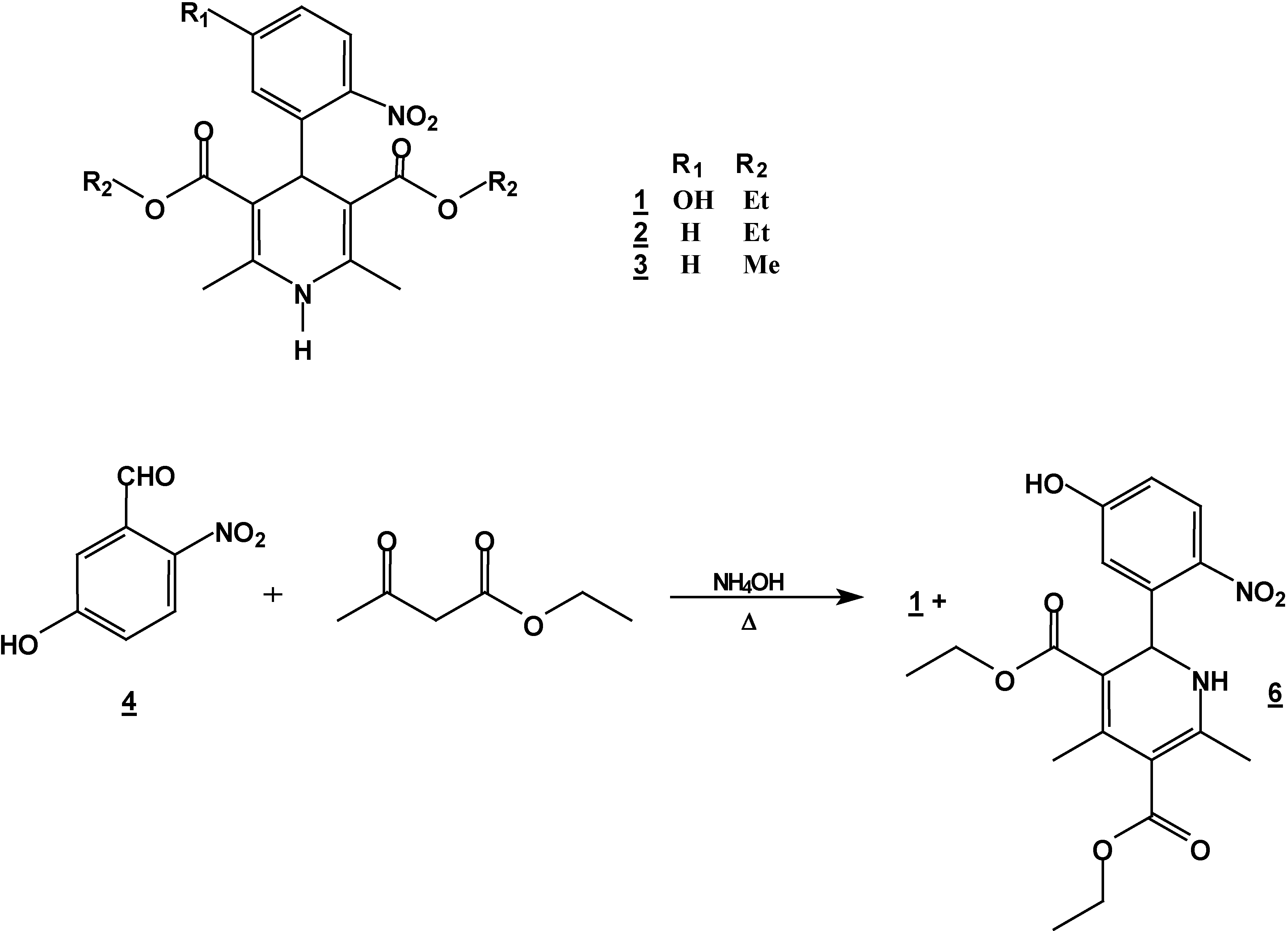
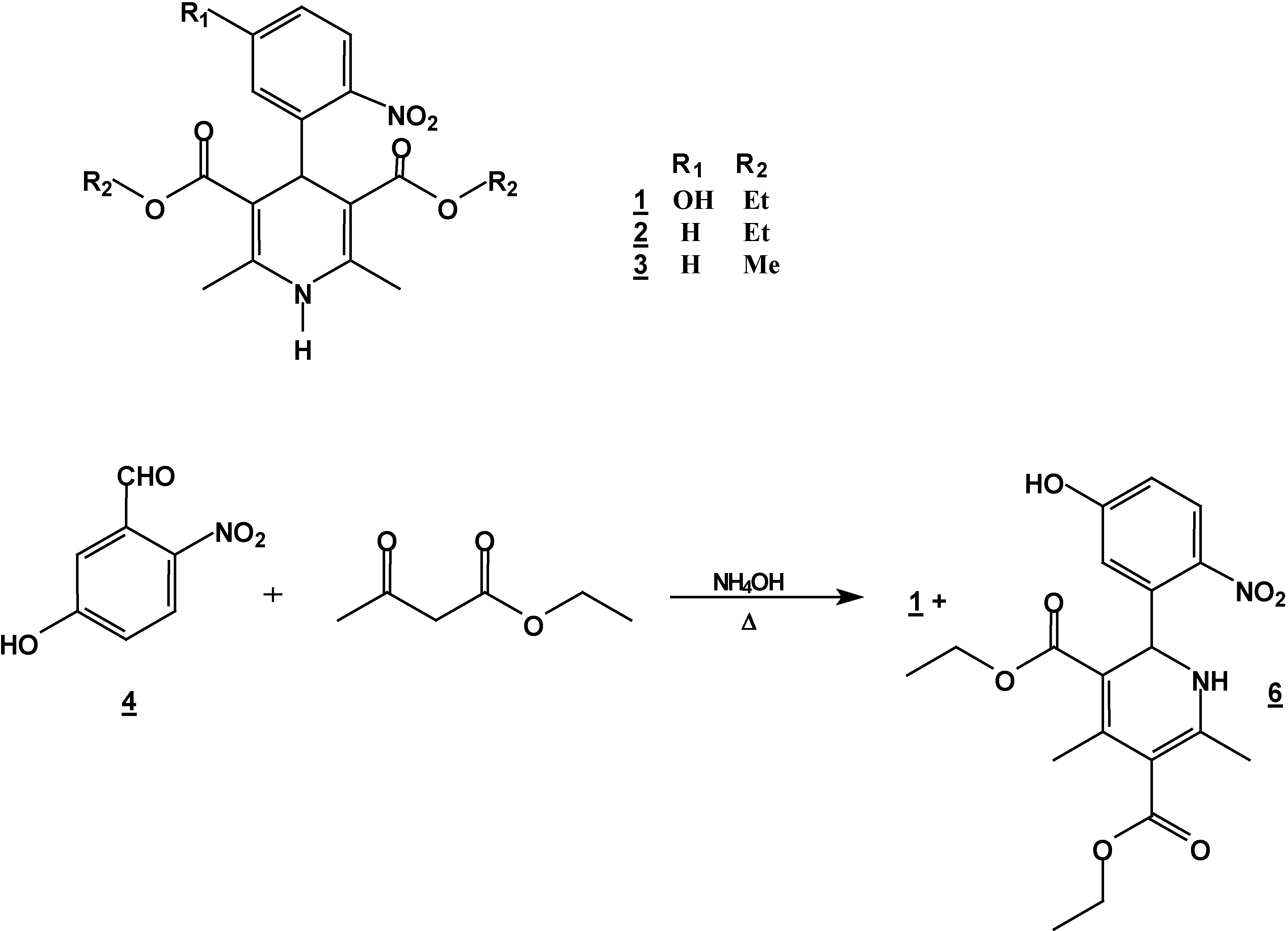
Synthesis of 4-(5-hydroxy-2-nitrophenyl)-1,4-dihydropyridine (1) + 2-(5-hydroxy-2-nitro-phenyl)-4,6-dimethyl-1, 2-dihydropyridine-3,5-dicarboxylic acid diethyl ester (6).
A mixture of 5 g (0.30 mole) of 5-hydroxy-2-nitrobenzaldehyde (5g, 0.30 mole), 7.8 g (0.60 mole) of ethyl acetoacetate (7.8g ,0.60 mole) and 30% 25 ml of ammoniaammonium hydroxide (25 mL)30%) in ethanol ( 250 mLl) was stirred under reflux for 2 hours. The solution was concentrated (rotatory evaporator) to afford a solid mixtureresidue. This mixture was then separated by column chromatography (silica gel, hexane/ ethyl acetate , gradient) into compounds 1 (5.57 g, 48 %) and 6 (2.32 g, 20 %); m.p. 206-208°C (hexane/ethyl acetate); IR (CHCl3 film) νmax/cm−1: 3346, 3240, 1701, 1656; 1H-NMR: 10.3 (s, 1H, OH), 7.96 (d, J=6.0Hz, 1H, H3*), 6.97 (d, J=6.0Hz 1H, H6*), 6.80 (dd, J=6.0, J=1.8 Hz, 1H, H4*), 6.52 (d, J=2.5 Hz, 1H, NH), 6.18 (d, J=2.5 Hz, 1H, H2), 4.20 (q, J=9.4 Hz, 2H, H10), 4.0 (q, J=9.4 Hz, 2H, H13), 2.51 (s, 3H, H7), 2.14 (s, 3H, H8), 1.28 (t, J=9.4 Hz, 3H, H14), 1.06 t, J=9.4 Hz, 3H, H11); 13C-NMR: 167.2 (C=O, C9), 165.0 (C=O,C12), 163 (C5*), 154.5 (C6), 148.3 (C4), 139.8 (C2*), 138.5 (C3), 127.0 (C3*), 116.4 (C6*), 114.7 (C4*), 107.0 (C1*), 102.2 (C5), 59.0 (C13), 58.8 (C10), 50.2 (C2), 20,8 (C7), 18.6 (C8), 13.8 (C14), 13.4 (C11); M.S. IE m/z (%)= 390 (15, M+), 373 (18), 345 (30), 299 (100).
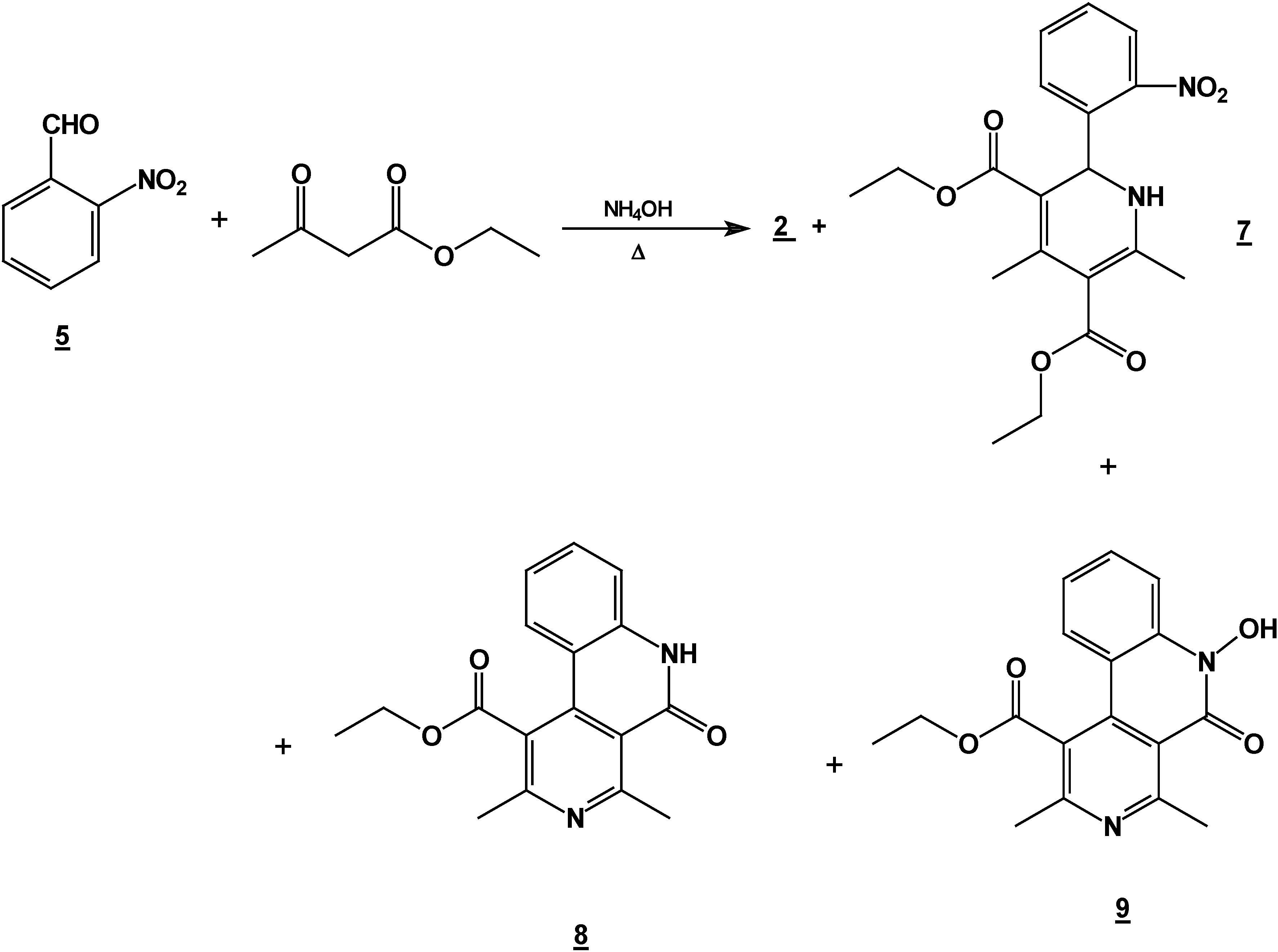
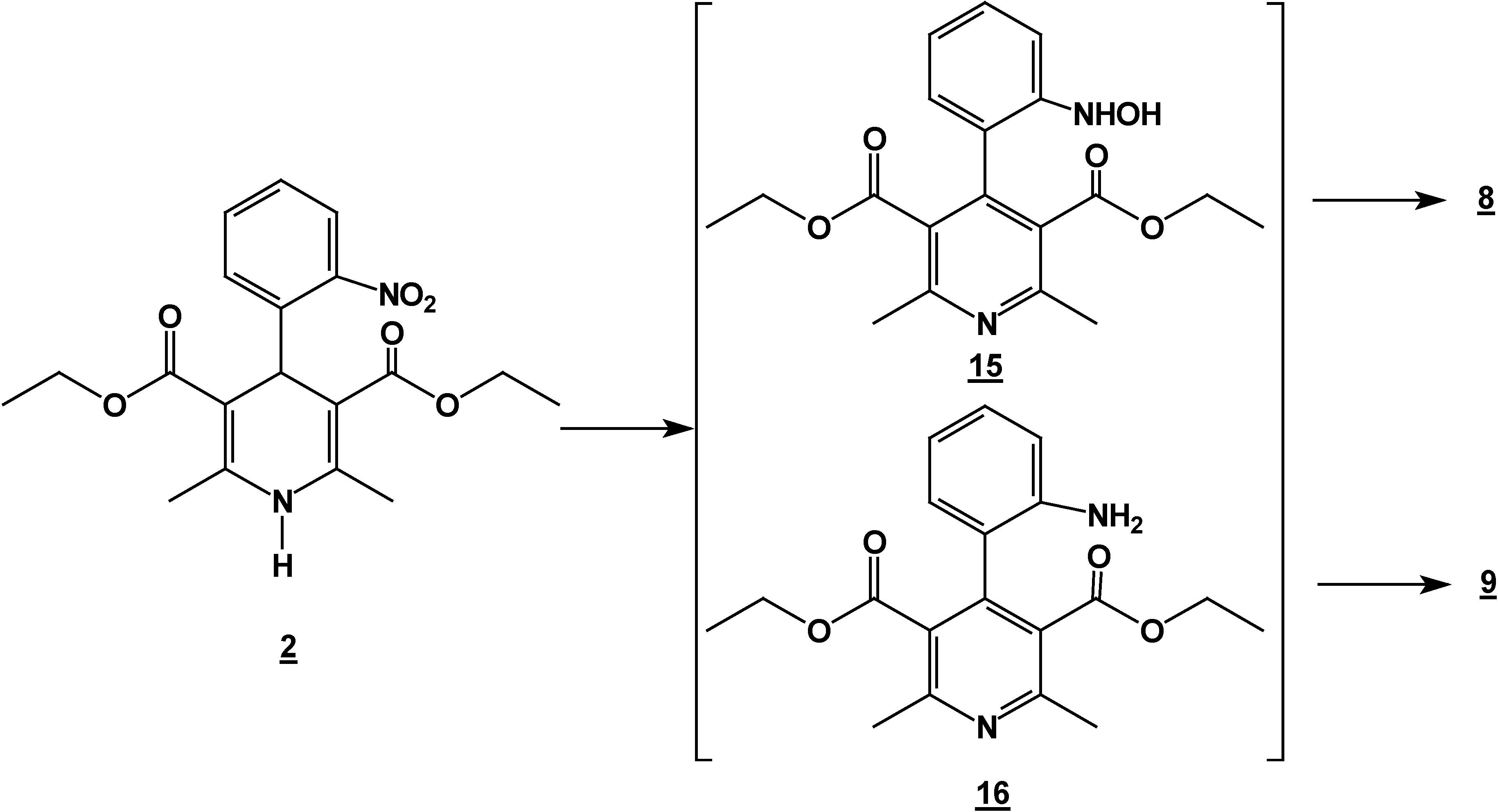
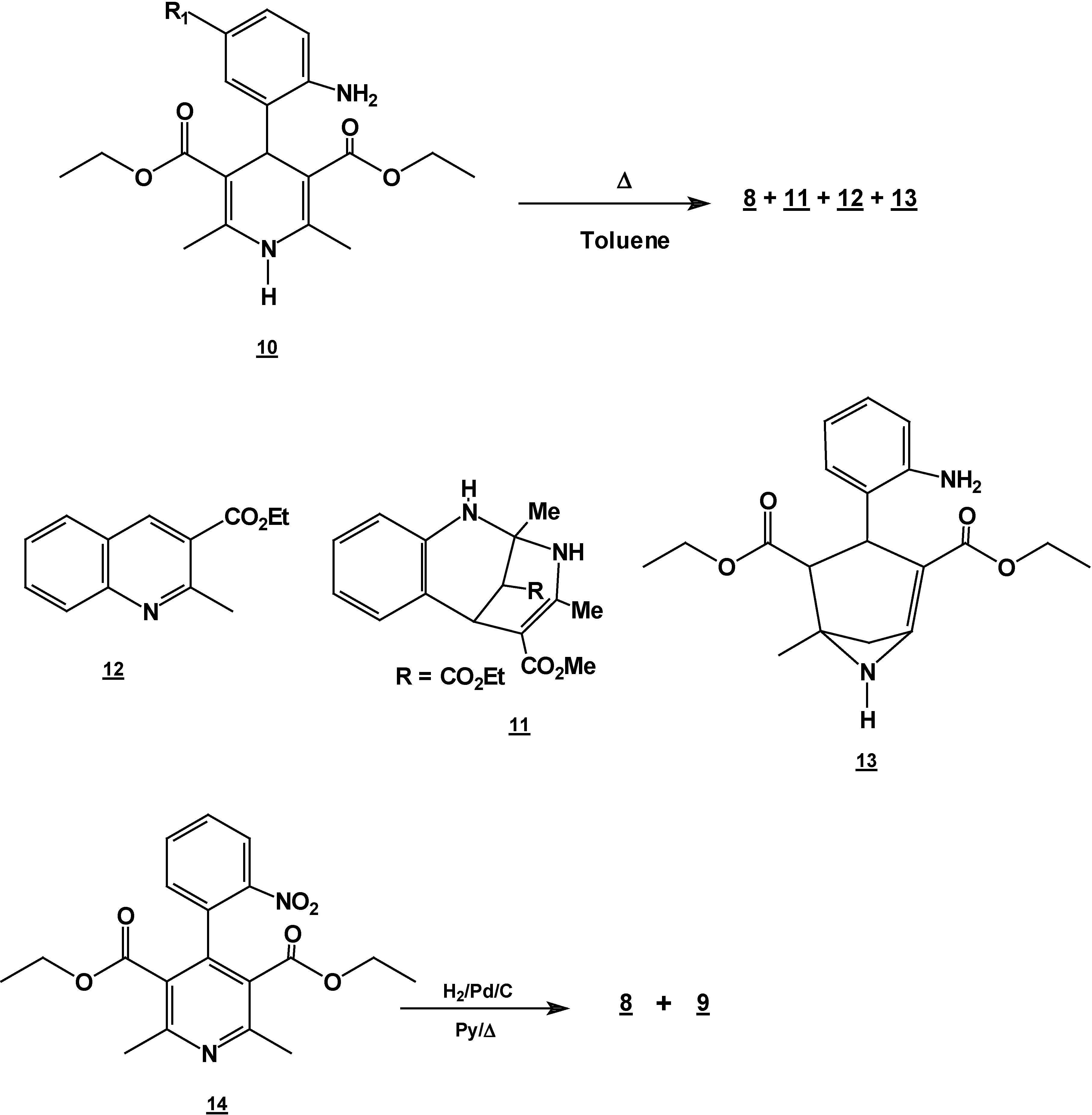
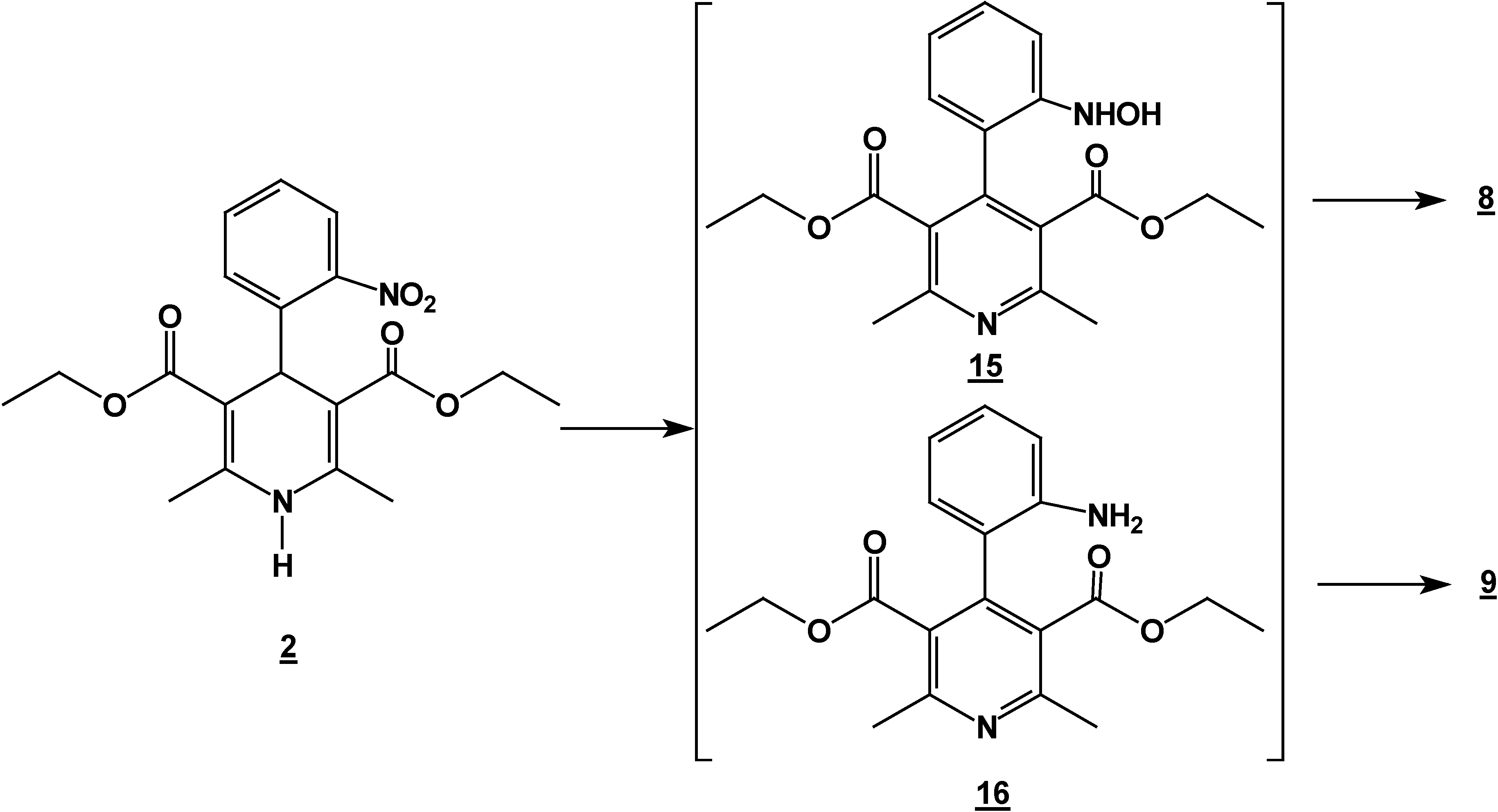
Synthesis of 2,6-Dimethyl 4-(2-nitro-phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (2) + 4,6-Ddimethyl 2-(2-nitro-phenyl)-1,2-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (7) + 5,6-Ddihydro-2,4-dimethyl-5-oxobenzo[c][2,7]naphthyridine-1-carboxylic acid ethyl ester (8) and 5,6-dDihydro-6-hydroxy-2,4-dimethyl-5-oxobenzo[c][2,7]naphthyridine-1-carboxylic acid ethyl ester (9).
A mixture of 10 g (0.066 mole) of 2-nitrobenzaldehyde (10g, 0.066 mole), 17.2 g (0.122 mole) of ethyl acetoacetate (17.2g, 0.122 mole) and 30 ml of 30% ammoniaammonium hydroxide (30%) mL) in ethanol ( 60 mLl) was stirred under reflux for 2 hours. The solution was concentrated (rotatory evaporator) to afford a solid mixture. This mixture was then separated by column chromatography (silica gel, hexane/ ethyl acetate gradient) into
2 [
8] (8.6 g, 35 %),
8 [
9] (2.94 g, 15 %),
9 [
9] (4.1 g, 20 %) and
7 (4.90 g, 20%), m.p. 158-160 °C; IR (CHCl
3 film) ν
max/cm
−1: 3450 (NH), 1710 (C=O), 1650 (C=O);
1H-NMR: 8.29-7.35 (m, Ar-HPh, 4H), 4.35 (q, J=7.4 Hz, 2H, H10), 4.09 (q, J=7.4 Hz, 2H, H9), 2.51 (s, 3H, H13), 2.21 (s, 3H, H14), 1.65 (s, 1H, NH), 1.35 (t, J=7.4 Hz, 3H, H11), 0.95 (t, J=7.4 Hz, 3H, H12);
13C-NMR: 166.3 (C=O, C7), 164.0 (C=O, C8), 122-140 (C-ArPh), 62.0, (C2), 61.0 (C9, C10), 31 (C13), 27.2 (C14), 14.2 (C11), 13.4 (C12).
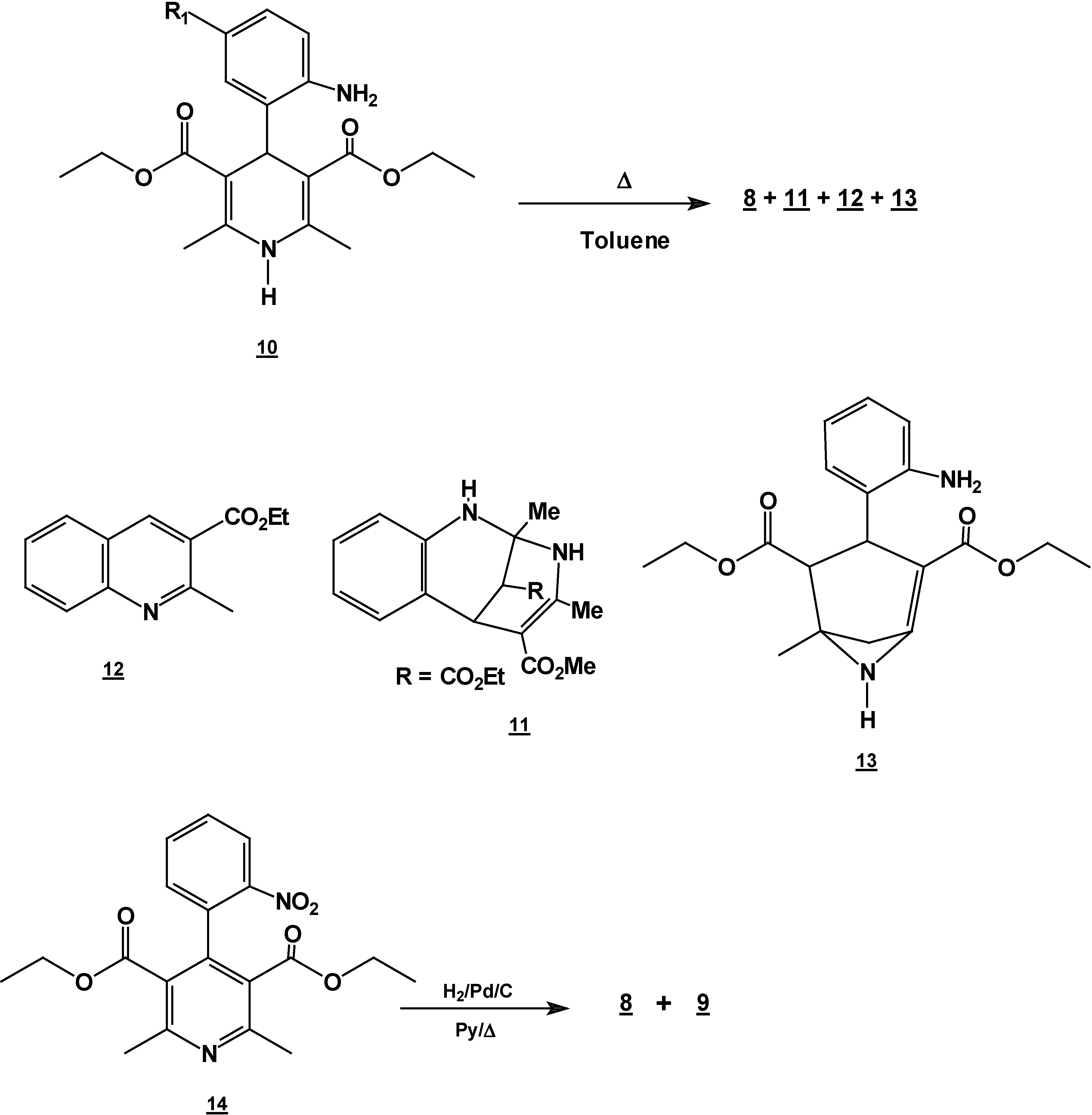
Reaction of 2,6-Dimethyl 4-(2-nitro-phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (2) with Tin (II) Chloride in Hydrochloric Acid.
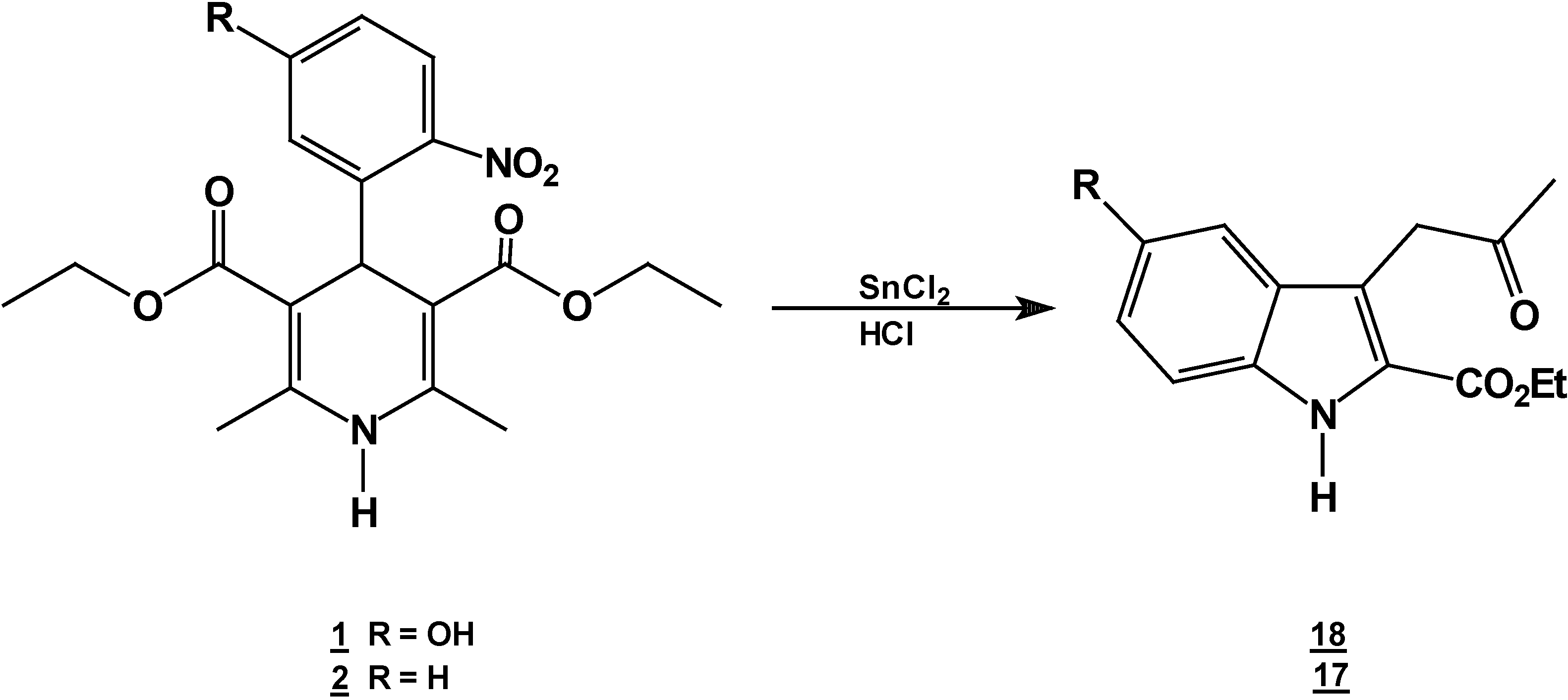
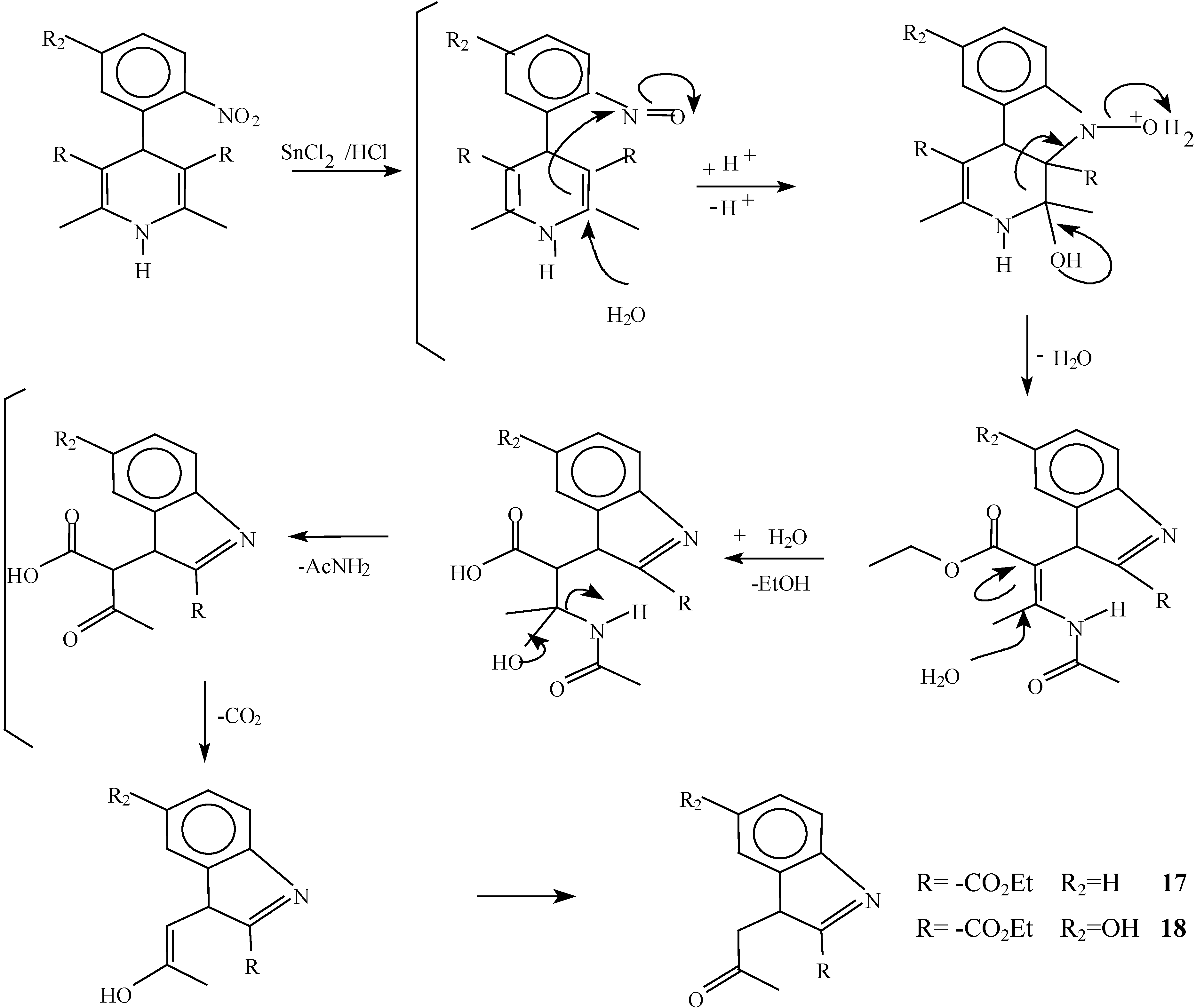
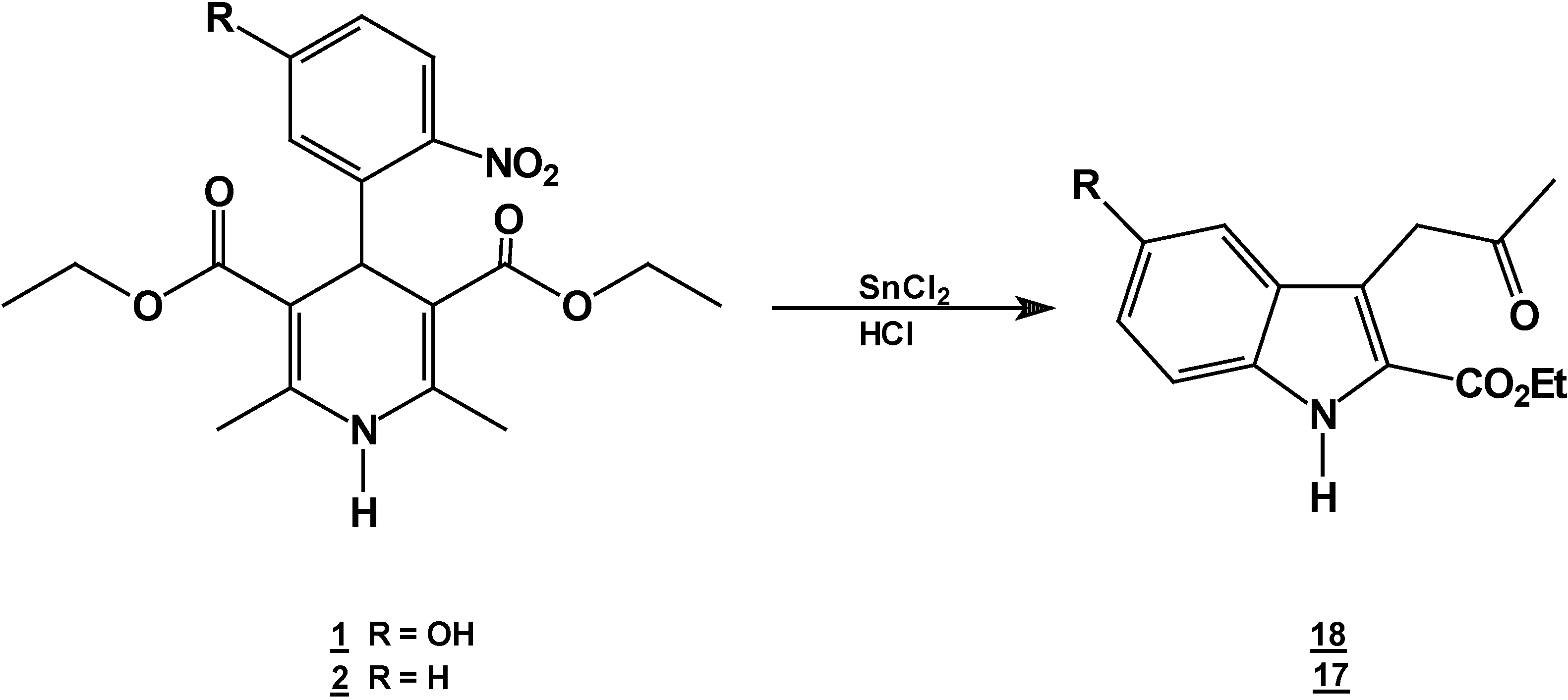
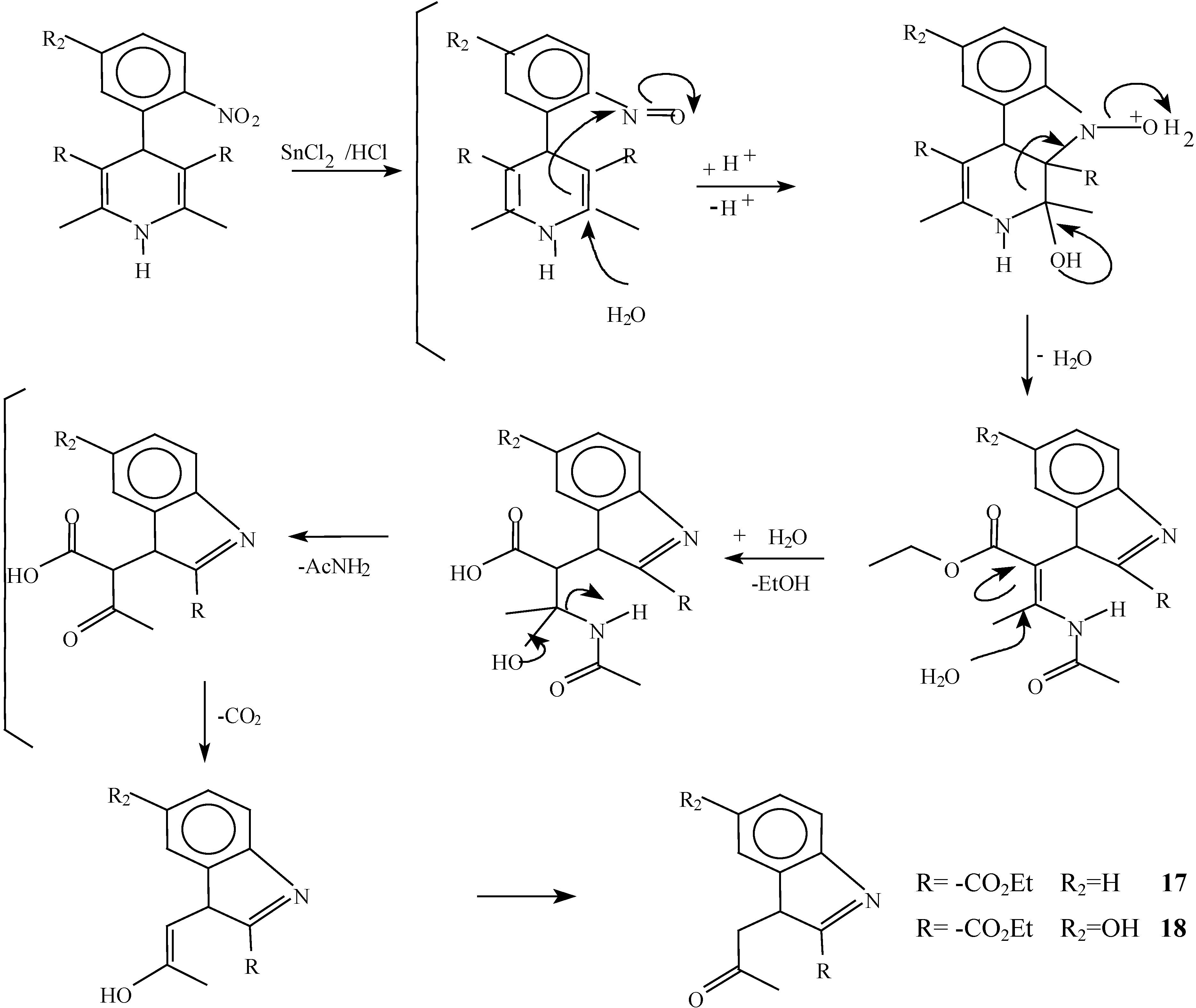
A solution of 1,4-dihydropyridine 2 (1.0 g, 0.0026 mol) in 25 ml of ethanol (25 mL) was added to a solution of stannoustin (II) chloride (5.4 g, 0.018 mole) in 30% 7 ml of hydrochloric acid (7 mL30%) at 5 °C. The reaction was allowed to warm to room temperature, and then heated to reflux for 5 hoursh.. The solution was neutralized with a 10% sodium bicarbonate solution and extracted with ethyl acetate (10 mLl × 3). The organic layer was dried (anhydrous sodium sulphatesulfate), filtered, and concentrated to afford crude product, which was purified by re crystallization from ethyl acetate/hexane to give 3-(2-oxopropilpropyl)-1H-indole-2-carboxilatecarboxylic acid ethyl ester (17) (0.21 g, 40%); m.p. 114-116 °C; IR (KBr pellet) νmax/cm−1: 3337 (N-H), 1724 (C=O), 1674 (C=O); 1H-NMR: 8.92 (sbr, 1H, N-H), 7.6 (ddd, J= 1.0 Hz, J= 8.0 Hz, 1H, H7), 7.38-7.10 (m, 3H, H4,H5, H6), 4.40 (q, J= 14.2 Hz, 2H, H9), 4.21 (s, 2H, H11), 2.18 (s, 3H, CH3), 1.41 (t, J= 14.2 Hz 3H, H10); 13C-NMR: 206.1 (C12), 161.8 (C8), 135.8 (Ca), 127.9 (Cb), 125.9 (C6), 124.2 (C2), 120.7 (C4), 120.5 (C5), 116.5 (C3), 111 (C7), 61.0 (C9), 40.3 (C11), 29.1 (C13), 14.2 (C10); MS m/z (%), 245 (50, M+), 202 (100 ) [ M-C2H3O], 156 ( 90) [M-C4H9O2]
Reaction of [2,6-Diethyl 4-(5-hydroxy-2-nitro-phenyl)-1,4-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (1) with Tin (II) Chloride in Hydrochloric Acid.
A solution of 1,4-dihydropyridine 1 (1.0 g, 0.0025 mol) in 25 ml of ethanol (25 mL) was added to a solution of stannoustin (II) chloride (5.4 g, 0.018 mole) in 30% 7 ml of hydrochloric acid (7mL) 30%) at 5 °C. The reaction was allowed to warm to room temperature, and then heated to reflux for 5 h. The solution was neutralized with a 10% sodium bicarbonate solution and extracted with ethyl acetate (10 mLl × 3). The organic layer was dried (anhydrous sodium sulfaphate), filtered, and concentrated to afford crude product, which was purified by recrystallization from ethyl acetate/hexane to afford 3-(2-oxopropyl)-5-hydroxy-1H-indole-2-carboxylic acid ethyl ester (18), m.p.Mp 154-156 °C, ; yield 0.313 g (56 %). IR (KBr pellet) νmax/cm−1: 3550 (OH), 3337 (N-H), 1720 (C=O), 1680 (C=O); 1H-NMR: 8.92 (sbr, 1H, N-H), 7.6 (ddd, J= 1.0 Hz, J= 8.0 Hz, 1H, H7), 7.38-7.10 (m, 3H, H4, H5, H6), 4.40 (q, J= 14.2 Hz, 2H, H9), 4.21 (s, 2H, H11), 2.18 (s, 3H, CH3), 1.41 (t, J= 14.2 Hz 3H, H10); 13C-NMR: 206.1 (C12), 161.8 (C8), 135.8 (Ca), 127.9 (Cb), 125.9 (C6), 124.2 (C2), 120.7 (C4), 120.5 (C5), 116.5 (C3), 111 (C7), 61.0 (C9), 40.3 (C11), 29.1 (C13), 14.2 (C10).
Reaction of [4,6-Dimethyl 2-(2-nitro-phenyl)-1,2-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (7) with Tin (II) Chloride in Hydrochloric Acid.
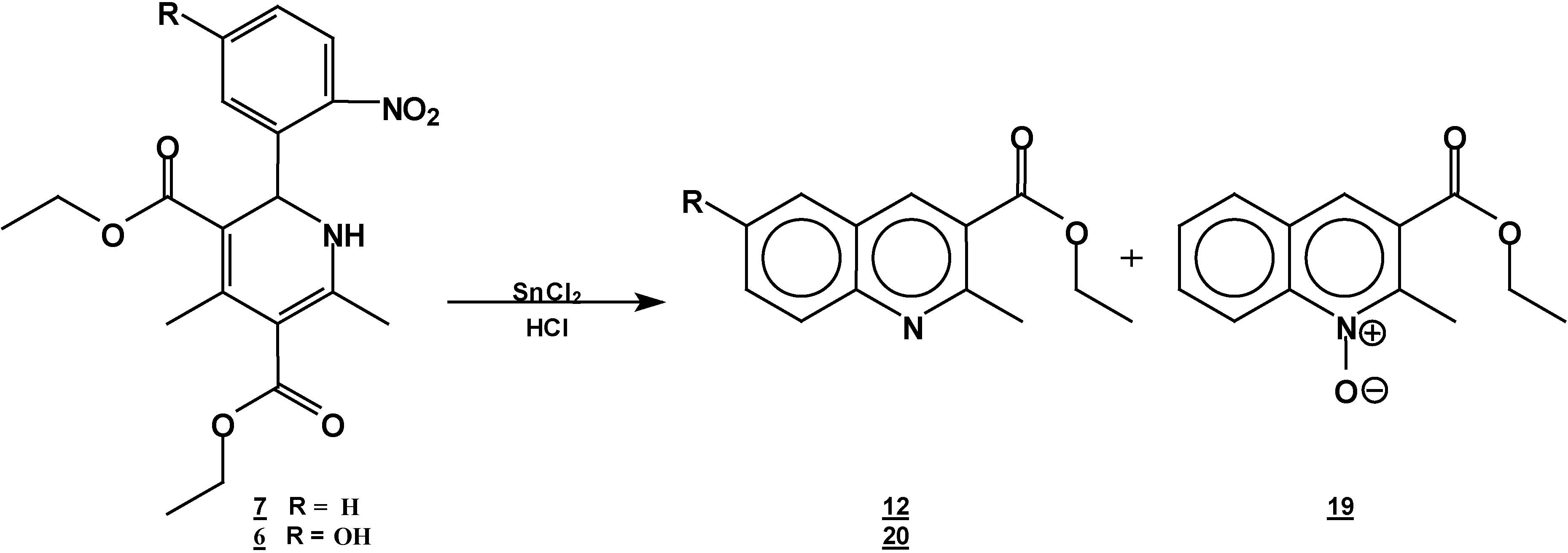
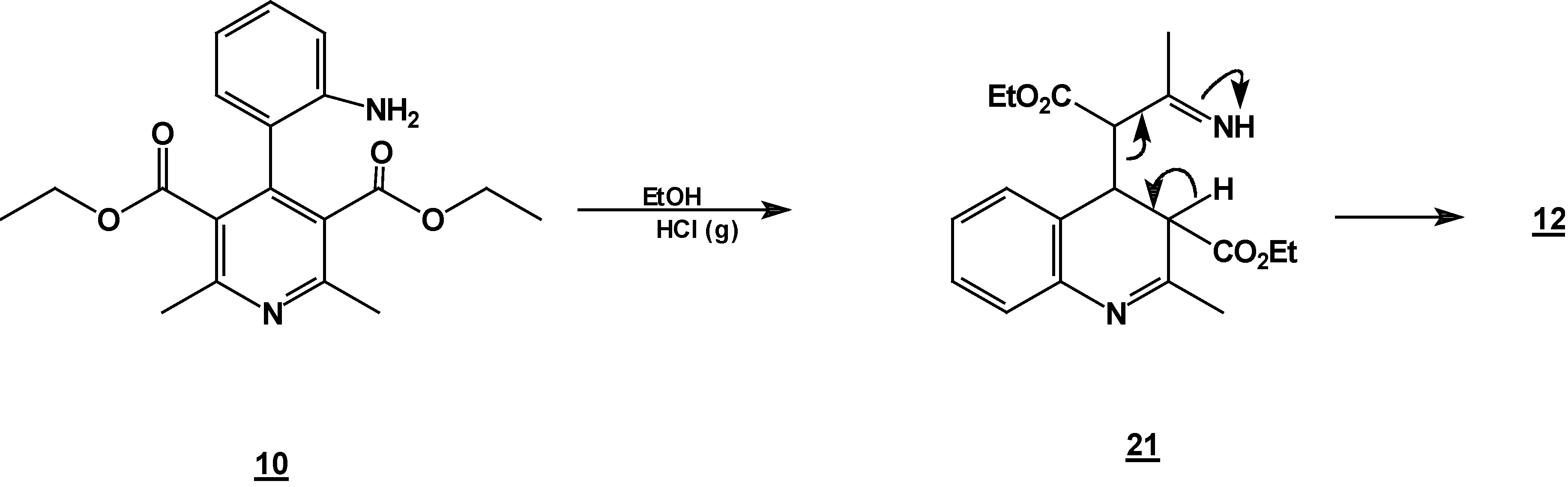
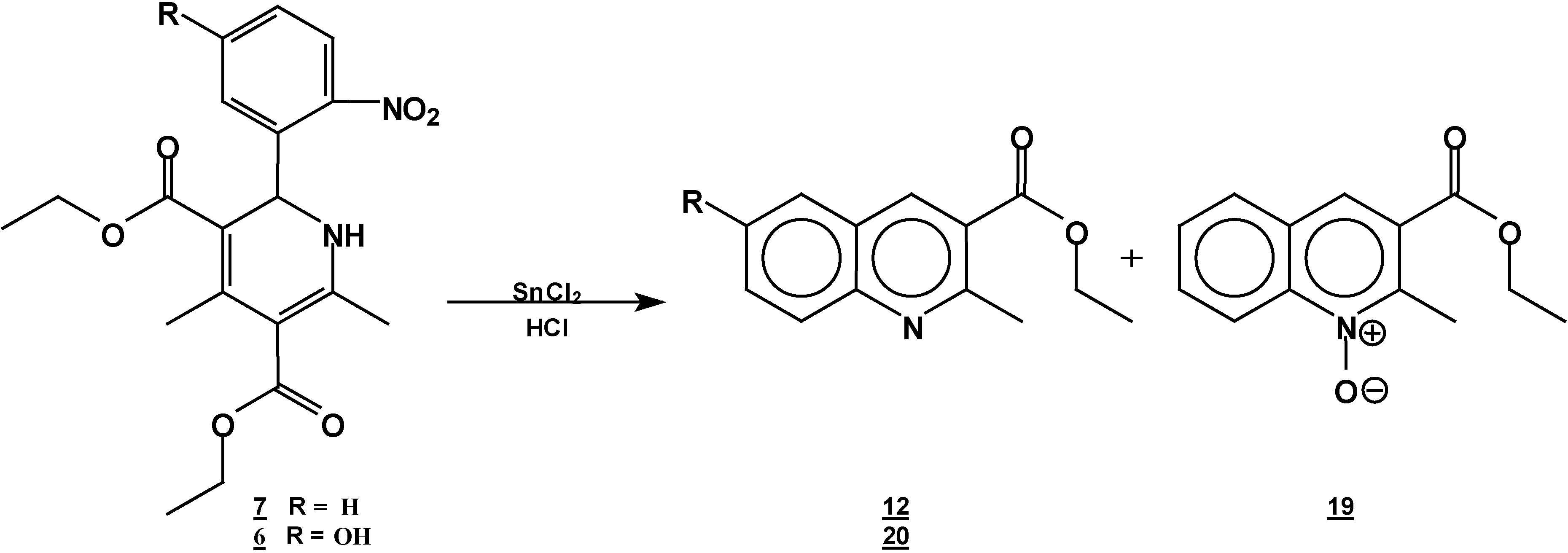
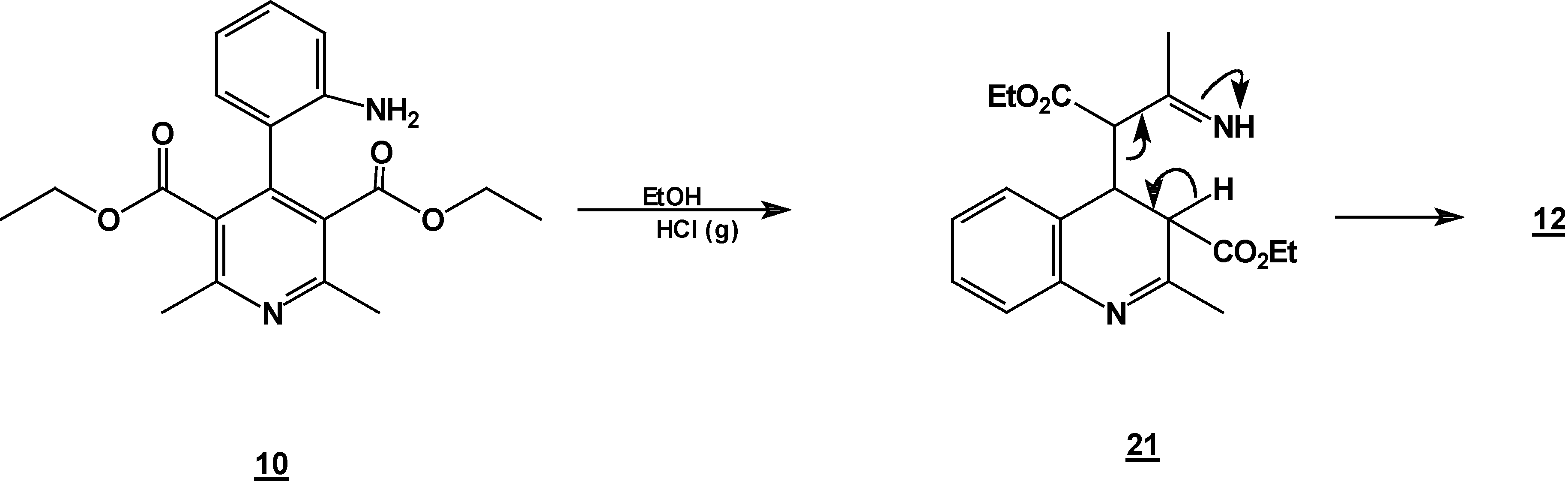
A solution of 1,2-dihydropyridine
7 (1.0 g, 0.0026 mol) in 25 ml of ethanol (25 mL) was added to a solution of stannoustin (II) chloride (5.4 g, 0.018 mole) in 30% hydrochloric acid (7 mL) at 5 °C. The reaction was allowed to w arm to room temperature, and then heated to reflux for 5 h. The solution was neutralized with a 10% sodium bicarbonate solution and extracted with ethyl acetate (10 ml × 3). The organic layer was dried (anhydrous sodium sulphate), filtered, and concentrated to afford crude product, which was purified by column chromatography and then rethey were recrystallized from ethyl acetate/hexane to obtain give
2-methyl- quinoline-3-carboxylic acid ethyl ester (
12), m.p. 74-76 °C (lit [
18,
19] 70-72 °C) and
6-hydroxy-2-methyl-oxyquinoline-3-carboxylic acid ethyl ester (
19, 0.3 g (54%); m.p. 98-100 °C; IR (KBr pellet ) ν
max/cm
−1): 1716 (C=O) .-;);
1H-NMR: 8.8 (d, J= 1.2 Hz, 1H, H8), 8.26 (s, 1H, H4), 7.9-7.6 (m, 3H, H5, H6, H7 ), 4.45 (q, J= 14.1 Hz, 2H, H11), 2.96 (s, 3H, H9), 1.46 (t, J= 14.1 Hz, 3H, H12);
13C-NMR: 165.3 (C=O, C10), 146.3 (C2), 142.3 (Ca), 132.3 (C4), 129.0 (C6), 128.4 (C7), 127.6 (C5), 127.0 (C3), 126.0 (Cb), 119.9 (C8), 62 (C11), 15.8 (C9), 14.2 (C12); MS
m/z (%), 231 (75, M
+), 214 (15) [M-OH], 186 (100) [M-45].
Reaction of 2-(5-Hydroxy-2-nitro-phenyl)-4,6-dimethyl-1,2-dihydro-pyridine-3,5-dicarboxylic acid diethyl ester (6) with Tin (II) Chloride in Hydrochloric Acid
A solution of 1,2-dihydropyridine 6 (1.0 g, 0.0025 mol) in ethanol (25 mL) was added to a solution of stannoustin (II) chloride (5.4 g, 0.018 mole) in 30% hydrochloric acid (7 mL) at 5 °C. The reaction was allowed to warm to room temperature, and then heated to reflux for 5 h. The solution was neutralized with a 10% sodium bicarbonate solution and extracted with ethyl acetate (10 mlL × 3). The organic layer was dried (anhydrous sodium sulphate), filtered, and concentrated to afford crude product, which was purified by column chromatography and then by rerecrystallization from hexane/ethyl acetate to give 6-hydroxy-2-methylquinoline-3-carboxylic acid ethyl ester (20), yield 0.35 g (60 %); m.p. 153-155 °C; IR, (KBr pellet) νmax/cm−1)= 3431, (OH), 1725 (C=O); 1H-NMR (CDCl3 +DMSO): 9.56 (s,1 1H, OH), 9.56 (s, 1H, H4), 7.8 (d, J=9 Hz, 1H, H8), 7.4 (dd, J=2.7 Hz, J=9 Hz, 1H, H7), 7.15 (d, J=2.7Hz, 1H, H5), 4.42, (q, J=14.3Hz, 2H, ester CH2 ester), 2.9 (s, 3H, CH3- 2), 1.45 (t, J=14.3Hz, 3H, CH3 ester CH3); 13C-NMR- (CDCl3 +DMSO): 166.1 (C=O, C10), 155 (C6), 154 (C2), 143 (Ca), 137.5 (C4), 128.9 (C8), 126.4 (C3), 123.7 (C7), 123.4 ( Cb), 108.5 (C5), 60.6 (C10), 24.6 (C12), 13.7 ( C11); MS m/z (%), 231 (100, M+), 216 [M-15] (4), 202 [M-C2H5] (20), 186 [M-OEt] (45).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}