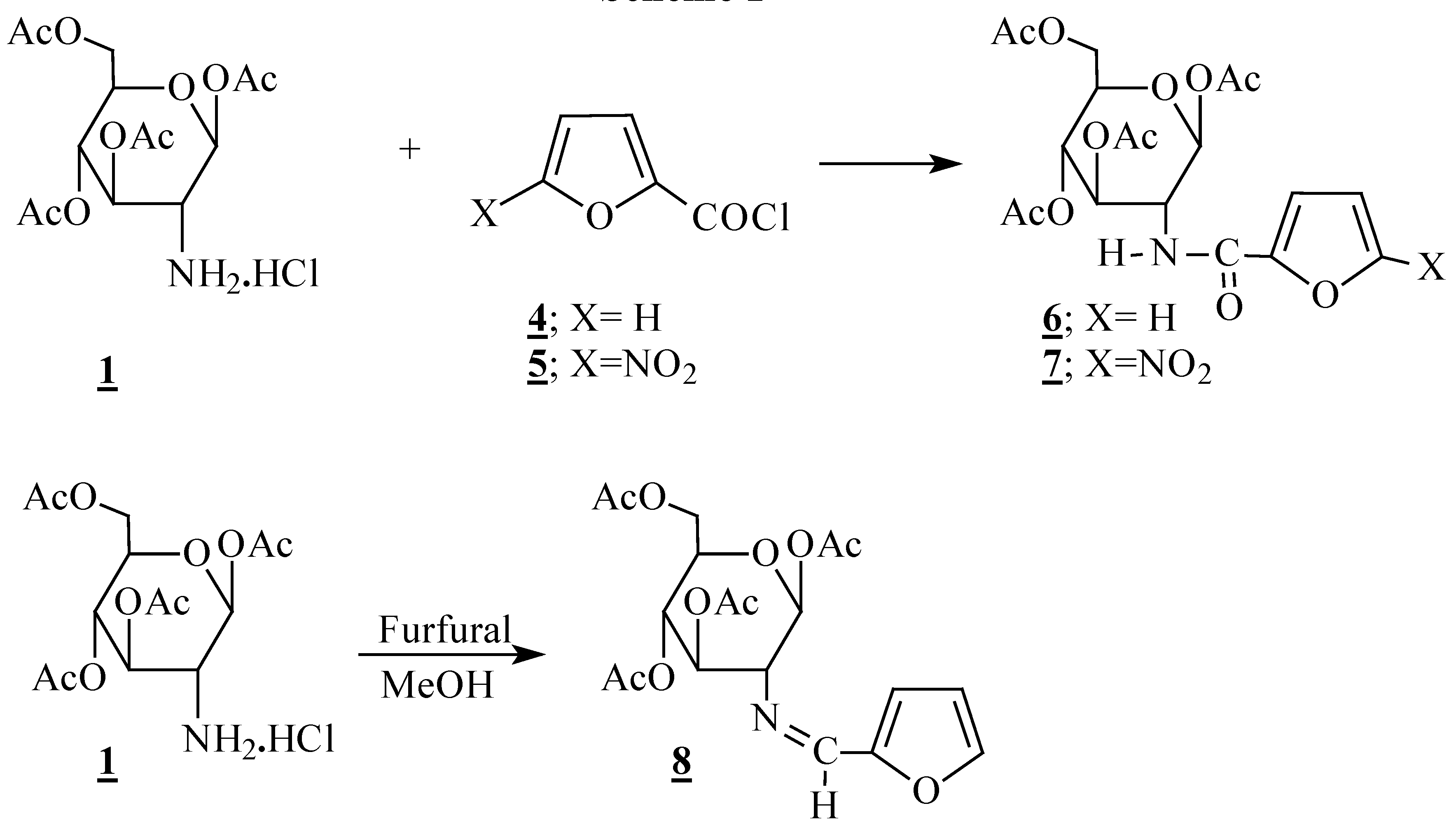
General method for condensation reactions: preparation of compounds 6, 7 and 9-12.
To a stirred solution of the amine (1 mmol) in anhydrous dichloromethane (10 mL) at 0 °C, were slowly added the acid chloride (1.5 mmol) and pyridine (3 mmol). The resulting mixture was stirred at 0 °C for 4 h and partitioned between water and dichloromethane. The organic phase was evaporated under vacuum to dryness and the residue was purified by column chromatography on silica gel, eluting with AcOEt/hexane.
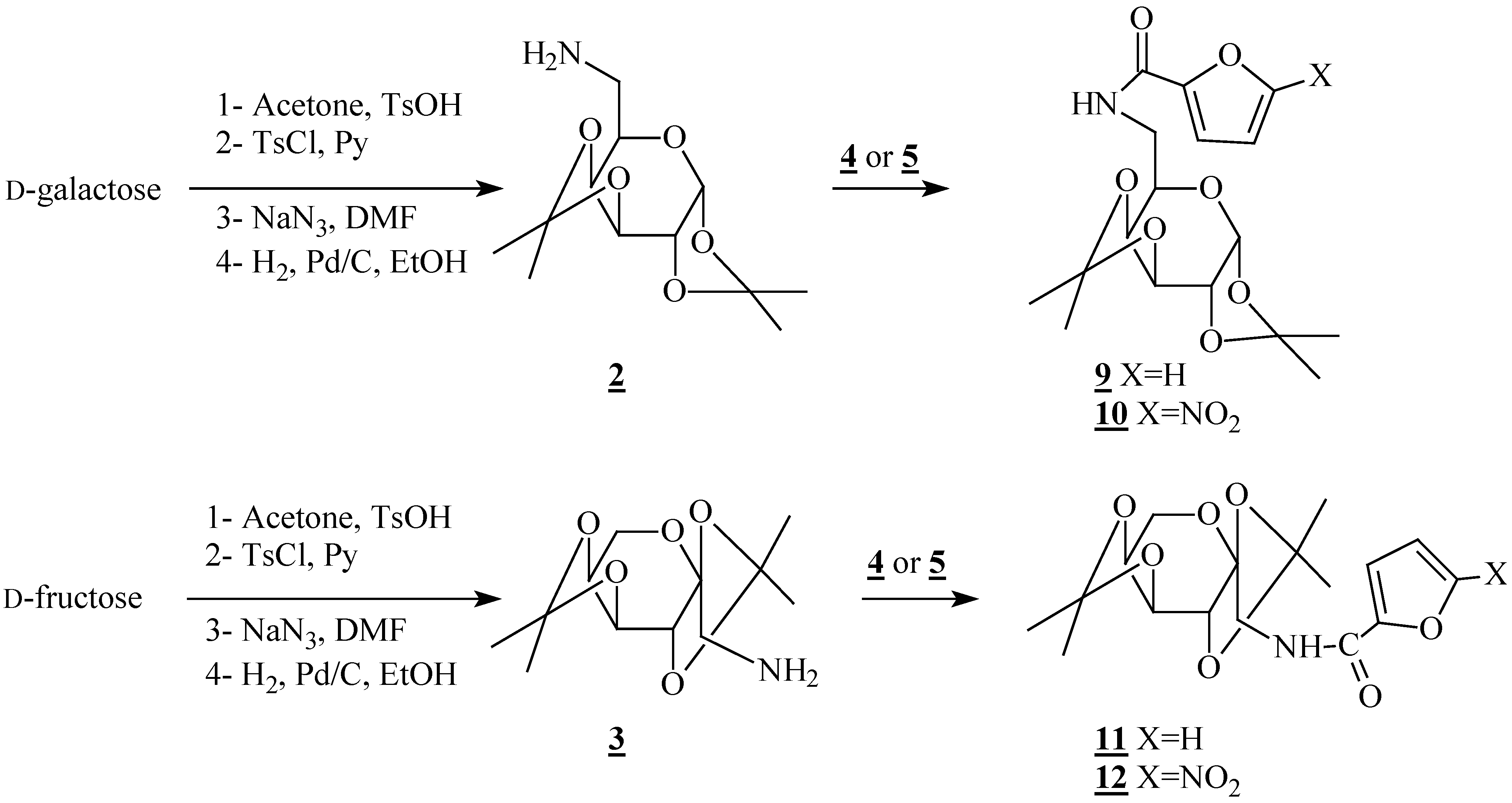
2-Deoxy-2-(2-furamido)-1,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (6): m p 208-210 °C; [α]D +55(c 1, CH2Cl2); IR (KBr) 1650 (NHC=O); 1750 (OC=O) cm-1; 1H-NMR (CDCl3) δ 1.99; 2.05; 2.08; 2.11; (4s, 12H, CH3); 3.87 (ddd, 1H, H5, J5-4=9.6 Hz; J5-6=4.6 Hz; J5-6'=2.2 Hz); 4.15 (dd, 1H, H6', J6'-6=12.5 Hz); 4.30 (dd, 1H, H6); 4.47 (q, 1H, H2); 5.19 (t, 1H, H4, J4-3=9.6 Hz); 5.30 (dd, 1H, H3, J3-2=10.4 Hz); 5.81 (d, 1H, H1, J1-2=8.7 Hz); 6.47 (d, 1H, NH, JNH-2=9.4 Hz); 6.49 (dd, 1H, H4a, J4a-3a=3.5 Hz, J4a-5a=1.2 Hz); 7.11 (dd, 1H, H3a); 7.43 (d, 1H, H5a); 13C-NMR (CDCl3) δ 20.56, 20.57, 20.72, 20.85 (CH3); 52.51 (C2); 61.68 (C6); 67.88; 72.43; 72.97 (C3, C4, C5); 92.63 (C1); 112.21; 115.16 (C3a, C4a); 144.56 (C5a); 146.93 (C2a); 158.10 (NHC=O); 169.29; 169.49; 170.67; 171.03 (OC=O); Anal. Calcd. for C19H23NO11: C, 51.70; H, 5.25; N, 3,17. found: C, 52.06; H, 4.92; N, 3.12.
2-Deoxy-2-(5-nitro-2-furamido)-1,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (7): m p 109-111 °C; [α]D +48 (c 0.7, DMSO); IR (KBr) 1660 (NHC=O); 1740 (OC=O) cm-1; 1H- NMR (CDCl3) δ 2.03; 2.09; 2.10 (3s, 12H, CH3); 3.98 (ddd, 1H, H5, J5-4=9.6 Hz; J5-6=4.9 Hz; J5-6'=2.3 Hz); 4.17 (dd, 1H, H6', J6'-6=12.5 Hz); 4.32 (dd, 1H, H6); 4.51 (q, 1H, H2); 5.20 (t, 1H, H4, J4-3 = J4-5=9.6 Hz); 5.50 (dd, 1H, H3, J3-2=10.4 Hz); 5.89 (d, 1H, H1, J1-2=8.7 Hz); 7.27 (d, 1H, H3a, J3a-4a=3.9 Hz); 7.30 (d, 1H, H4a); 7.41 (d, 1H, NH, JNH-2=9.6 Hz); 13C-NMR (CDCl3) δ 20.49, 20.59, 20.67, 20.86 (CH3); 52.58 (C2); 61.79 (C6); 68.11; 72.47; 72.81 (C3, C4, C5); 91.97 (C1); 112.31; 116.70 (C3a, C4a); 147.15; 151.19 (C2a, C5a); 156.30 (NHC=O); 169.28; 169.36; 170.64; 171.46 (OC=O); Anal. Calcd. for C19H22N2O13: C, 46.92; H, 4.56; N, 5,76. found: C, 46.98; H, 4.79; N, 5.49.
6-Deoxy-6-(2-furamido)-1,2;3,4-di-O-isopropylidene-α-D-galacto-pyranoside (9): oil; [α]D -35 (c 1, CH2Cl2); IR (KBr) 1650 (C=O) cm-1; 1H-NMR (CDCl3) δ 1.27; 1.32; 1.48 (3s, 12H, CH3); 3.42 (ddd, 1H, H6, J6-6'=14 Hz; J6-5=4 Hz); 3.89 (ddd, 1H, H6', J6'-5=4 Hz); 4.00 (ddd, 1H, H5, J5-4=1.8 Hz); 4.27 (dd, 1H, H4, J4-3=8.0 Hz); 4.32 (dd, 1H, H2, J2-1=5.0 Hz, J 2-3=2.4 Hz); 4.62 (dd, 1H, H3); 5.54 (d,1H, H1); 6.48 (dd, 1H, H4a, J4a-3a=3.5 Hz, J4a-5a=1.8 Hz); 6.80 (sl, 1H, NH); 7.10 (d, 1H, H3a); 7.43 (d, 1H, H5a); 13C-NMR (CDCl3) δ 24.33; 24.94; 25.93; 26.01 (CH3); 39.65 (C6); 66.27; 70.53; 70.82; 71.79 (C2, C3, C4, C5); 96.29 (C1); 108.81, 109.48 [C(CH3)2]; 111.95; 114.01 (C3a, C4a); 143.86 (C5a); 147.97 (C2a); 158.58 (C=O); Anal. Calcd. for C17H23NO7: C, 57.78; H, 6.56; N, 3,96. found: C, 57.67; H, 6.86; N, 3.78.
6-Deoxy-6-(5-nitro-2-furamido)-1,2;3,4-di-O-isopropylidene-α-D-galactopyranoside (10): oil; IR (KBr) 1650 (C=O) cm-1; 1H-NMR (CDCl3) δ 1.32; 1.38; 1.49; 1.54 (4s, 12H, CH3); 3.46 (ddd, 1H, H6, J6-6'=13.8 Hz; J6-5=4.2 Hz); 3.97 (ddd, 1H, H6', J6'-5=4.2 Hz); 4.02 (ddd, 1H, H5, J5-4=1.7 Hz); 4.30 (dd, 1H, H4, J4-3=7.9 Hz); 4.33 (dd, 1H, H2, J2-1=5.0 Hz, J2-3=2.4 Hz); 4.64 (dd, 1H, H3); 5.55 (d,1H, H1); 7.30 (sl, 1H, NH); 7.24; 7.34 (2d, 2H, H3a, H4a, J3a-4a=3.7 Hz); 13C-NMR (CDCl3) δ 24.30; 24.93; 25.96; 26.05 (CH3); 40.60 (C6); 65.23; 70.47; 70.94; 72.35 (C2, C3, C4, C5); 96.42 (C1); 108.87; 109.79 [C(CH3)2]; 112.36; 115.76 (C3a, C4a); 148.18; 151.20 (C2a, C5a); 156.56 (C=O); Anal. Calcd. for C17H22N2O9: C, 51.26; H, 5.57; N, 7.03. found: C, 51.53; H, 5.87; N, 6.67.
1-Deoxy-1-(2-furamido)-2,3;4,5-di-O-isopropylidene-β-D-fructopyranoside (11): oil; [α]D -16 (c 1.3, CH2Cl2); IR (KBr) 1640 (C=O) cm-1; 1H-NMR (CDCl3) δ 1.33; 1.41; 1.43; 1.54 (4s, 12H, CH3); 3.72 (dd, 1H, H1, J1-NH=6.0 Hz; J1-1'=14 Hz); 3.76 (d, 1H, H6, J6-6'=13.2 Hz); 3.85 (dd, 1H, H1', J1'-NH=6.0 Hz); 3.90 (dd, 1H, H6'); 4.22 (dd, 1H, H5, J5-4=7.7 Hz; J5-6'=2.1 Hz); 4.33 (d, 1H, H3, J3-4=2.6 Hz); 4.57 (d, 1H, H4); 6.49 (dd, 1H, H4a, J4a-3a=3.5 Hz, J4a-5a=1.8 Hz); 6.84 (sl, 1H, NH); 7.12 (dd, 1H, H3a); 7.42 (d, 1H, H5a); 13C-NMR (CDCl3) δ 23.85, 24.95, 25.69, 26.20 (CH3); 46.06 (C1); 61.48 (C6); 70.24; 70.45; 71.91 (C3, C4, C5); 102.69 (C2); 108.39; 109.01 [C(CH3)2]; 112.13; 114.44 (C3a, C4a); 143.66 (C5a); 147.89 (C2a); 158.52 (C=O); Anal. Calcd. for C17H23NO7: C, 57.78; H, 6.56; N, 3.96. found: C, 57.91; H, 6.41; N, 3.75.
1-Deoxy-1-(5-nitro-2-furamido)-2,3;4,5-di-O-isopropylidene-β-D-fructopyranoside (12): m p 159-162 °C; [α]D -27 (c 0.5, DMSO); IR (KBr) 1650 (C=O) cm-1; 1H-NMR (CDCl3) δ 1.37; 1.42; 1.48; 1.55 (4s, 12H, CH3); 3.72 (dd, 1H, H1, J1-NH=5.4 Hz; J1-1'=14 Hz); 3.77 (d, 1H, H6, J6-6'=13.1Hz); 3.86 (dd, 1H, H1', J1'-NH=5.8 Hz); 3.90 (dd, 1H, H6'); 4.24 (dd, 1H, H5, J5-4=7.7 Hz; J5-6= 2.0 Hz); 4.31 (d, 1H, H3, J3-4= 2.7 Hz); 4.60 (dd, 1H, H4); 7.16 (sl, 1H, NH); 7,28, 7.37 (2d, 2H, H3a, H4a, J3a-4a=3.7 Hz; 13C-NMR (CDCl3) δ 24.05, 24.86, 25.75, 26.13 (CH3); 46.79 (C1); 61.74 (C6); 70.22; 70.54; 71.91 (C3, C4, C5); 102.30 (C2); 108.77; 109.42 [C(CH3)2]; 112.38; 116.20 (C3a, C4a); 148.00; 151.20 (C2a, C5a); 156.50 (C=O); Anal. Calcd. for C17H22N2O9: C, 51.26; H, 5.57; N, 7.03. found: C, 51.46; H, 5.45; N, 6.85.
2-Deoxy-2-(furfural)-imino-1,3,4,6-tetra-O-acetyl-β-D-glucopyranoside (8). To a stirred solution of hydrochloride 1 (1 mmol) in anhydrous methanol (10 mL) at room temperature was added sodium bicarbonate (1.2 mmol) and distilled furfural (2 mmol). The mixture was heated at reflux for 12 h and the solvent was removed. The residue was redissolved in dichloromethane and filtered on celite. The solvent was removed in vacuo and the residue was crystallized from AcOEt/hexane, yielding 32% of 8. Attempts to purify compound 8 by chromatography led to decomposition of the product. m p 114-116 °C; IR (KBr) 1640 (C=N); 1740 (C=O) cm-1; 1H-NMR (CDCl3) δ 1.92; 2.03; 2.04; 2.09 (4s, 12H, CH3); 3.43 (dd, 1H, H2, J2-1=8.4 Hz; J2-3=9.6 Hz); 3.96 (ddd, 1H, H5, J5-4=9.6 Hz; J5-6=4.5 Hz; J5-6'=2.2 Hz); 4.12 (dd, 1H, H6', J6'-6=12.5 Hz); 4.37 (dd, 1H, H6); 5.13 (t, 1H, H4, J4-3=9.6 Hz); 5.43 (t, 1H, H3); 5.94 (d, 1H, H1); 6.50 (dd, 1H, H4a, J4a-3a=3.5 Hz; J4a-5a=1.8 Hz); 6.84 (d, 1H, H3a); 7.54 (d, 1H, H5a); 8.04 (s, 1H, HC=N); 13C-NMR (CDCl3) δ 20.48; 20.58; 20.63; 20.77 (CH3); 61.73 (C6); 68.07; 72.64; 73.08; 73.19 (C2, C3, C4, C5); 93.08 (C1); 111.86, 115.89 (C3a, C4a); 145.67 (C5a); 150.70 (C2a); 153.2 (C=N); 168.50; 169.40; 169.90; 170.61 (C=O);
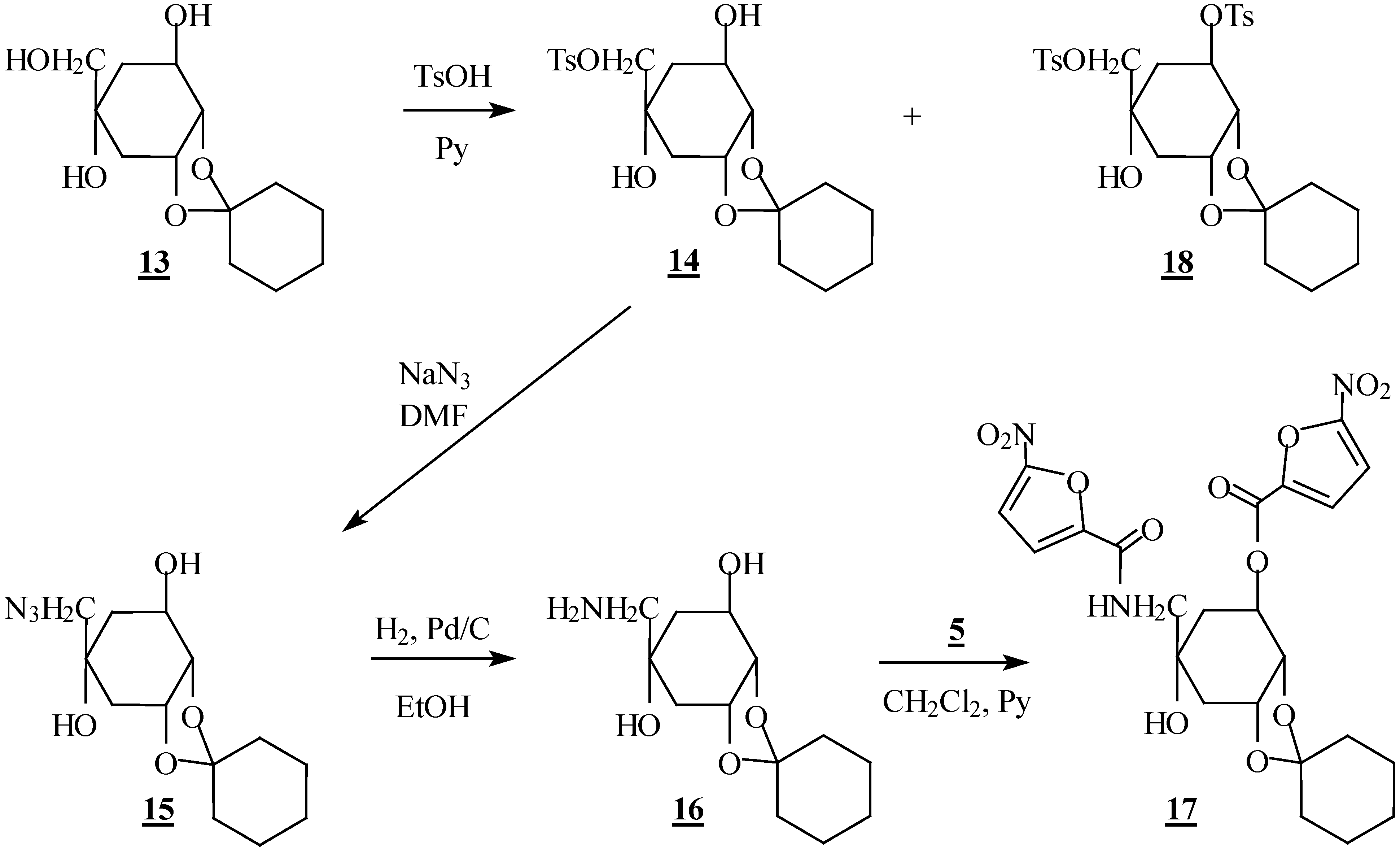
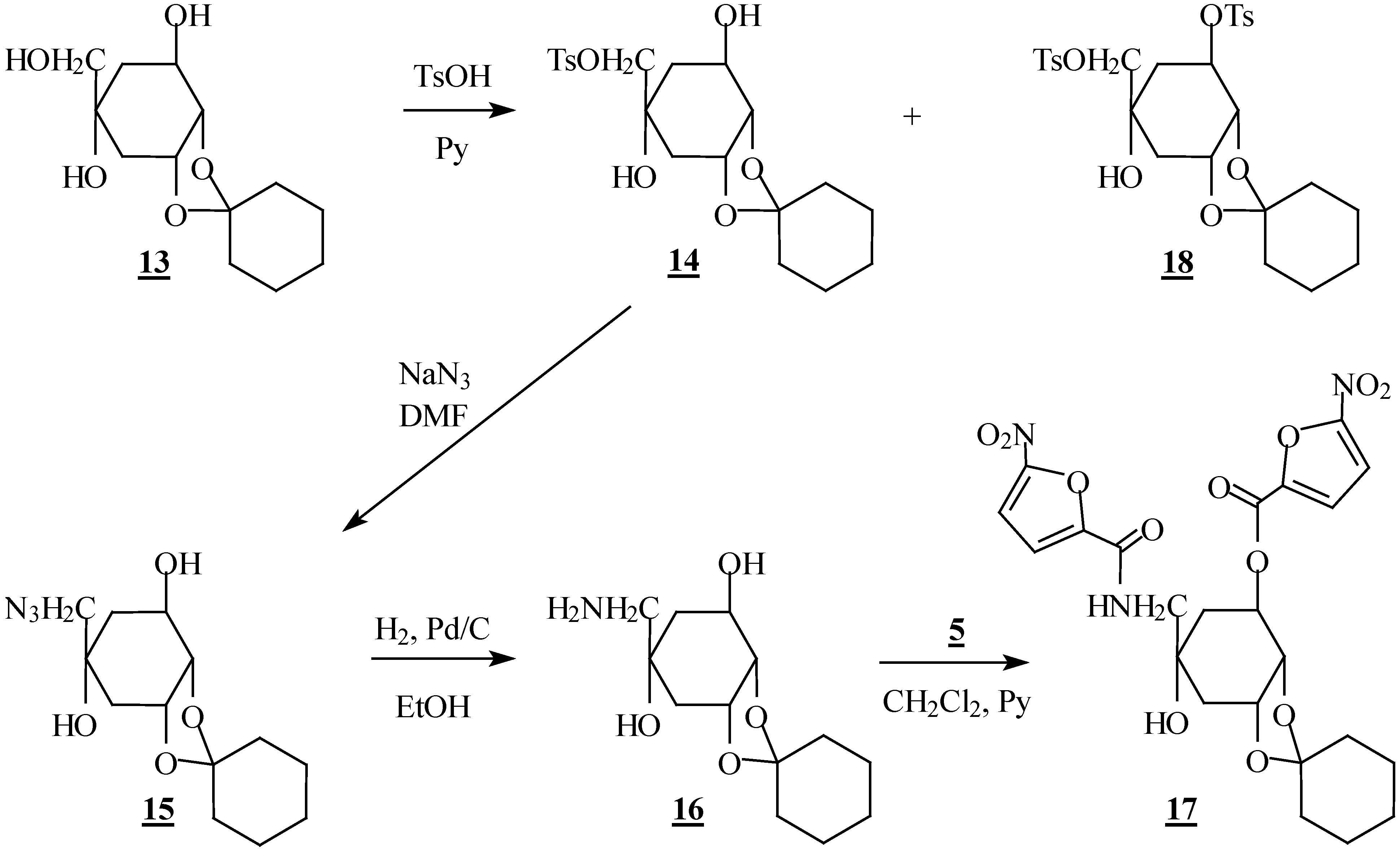
Preparation of compounds 14 and 18
To a solution of compound 13 (2.58 g, 10 mmol) in pyridine (20 mL) at 0 °C was added, in small portions, p-toluenesulfonyl chloride (2.28 g, 12 mmol). The mixture was stirred for 24 h at room temperature and extracted with AcOEt. The organic phase was concentrated in vacuo and the residue was chromatographed on silica gel to give the desired compound 14 and the ditosylate 18 in 60% and 15% yield, respectively.
(1R,2S,3R,5R)-1,2-O-cyclohexylidene-5-C-[(O-tosyl)-hydroxymethyl]-cyclohexane-1,2,3,5-tetrol (14): m p 134-136 °C; [α]D -16 (c 0.5, DMSO); IR (KBr) 1350 (ROSO2R) cm-1; 1H-NMR (CDCl3) δ 1.60 (m, 11H, cyclohex., H6); 1.86 (ddd, 1H, H6', J6'-6=13.5 Hz); 1.90 (dd, 1H, H4, J4-4'=15.4 Hz); 2.18 (d, 1H, OH); 2.20 (dt, 1H, H4'); 2.45 (s, 3H, CH3); 3.24 (s, 1H, OH); 3.82 (d, 1H, H7); 3.86 (d, 1H, H7', J7-7'=9.7 Hz); 3.90 (t, 1H, H2, J2-1=J2-3=6.0 Hz); 4.09 (m, 1H, H1); 4.42 (m, 1H, H3); 7.35; 7.80 (2d, 4H, Ph); 13C-NMR (CDCl3) δ 25.65 (CH3); 23.61; 23.97; 24.87; 33.15; 34.81; 38.05; 38.16 (cyclohex., C4, C6); 69.21; 73.71; 79.79; (C1, C2, C3); 71.13 (C5); 75.36 (C7); 110.05 (OCO); 127.98; 129.92; 132.63; 145.02 (Ph). Anal. Calcd. for C20H28O7S: C, 58.24; H, 6.84. found: C, 58.17; H, 6.82.
(1R,2S,3R,5R)-1,2-O-cyclohexylidene-3-O-tosyl-5-C-[(O-tosyl)-hydroxy-methyl]-cyclohexane-1,2,3,5-tetrol (
18): m p 124-126 °C (lit [
13] m p 123-125 °C); IR (KBr) 1350 (ROSO
2R) cm
-1;
1H-NMR (CDCl
3) δ 1.50 (m, 10H, cyclohex.); 1.89 (dd, 1H, H4, J
4-4'=15.7 Hz); 1.99 (dt, 1H, H6, J
6-6'=13.6 Hz; J
6-1=J
6-5=3.2 Hz); 2.07(ddd, 1H, H6'); 2.20 (dt, 1H, H4'); 2.46; 2.48 (2s, 6H, CH
3); 3.20 (sl, 1H, OH); 3.77 (d, 1H, H7); 3.83 (d, 1H, H7', J
7'-7=9.7 Hz); 4.06 (t, 1H, H2, J
2-1=J
2-3=6.0 Hz); 4.44 (m, 1H, H1); 4.75 (ddd, 1H, H3); 7.34; 7.37; 7.80; 7.82 (4d, 8H, Ph); );
13C-NMR (CDCl
3) δ 21.64; 21.73 (CH
3); 23.48; 23.83; 24.83; 32.82; 34.53; 36.62; 37.43 (cyclohex., C4, C6); 73.62; 76.00; 79.77 (C1, C2, C3); 70.86; 75.00 (C5, C7); 110.35 (O
CO); 128.00; 128.15; 129.77; 130.01; 132.49; 133.60; 144.78; 145.20 (Ph).
(1R,2S,3R,5R)-1,2-O-cyclohexylidene-5-C-azidomethyl-cyclohexane-1,2,3,5-tetrol (15). Sodium azide (0.65 g, 10 mmol) was added to a solution of the tosylate 14 (2.06 g, 5 mmol) in DMF (20 mL). The mixture was stirred overnight at 140 °C and extracted with AcOEt. The organic phase was concentrated in vacuo and the residue was chromatographed on silica gel to afford the azide derivative 15 in 95% yield; m p 100-102 °C; IR (KBr) 2100 (N3) cm-1; 1H-NMR (CDCl3) δ 1.60 (m, 11H, cyclohex., H6); 1.80 (dd, 1H, H4, J4-4'=15.5 Hz); 2.02 (ddd, 1H, H6', J6'-6=13.4 Hz); 2.30 (dt, 1H, H4'); 2.59 (d, 1H, OH); 3.13 (d,1H, H7, J7-7'=12.3 Hz); 3.28 (d, 1H, H7'); 3.37 (s, 1H, OH); 3.93 (t, 1H, H2, J2-1=J2.3=6.0 Hz); 4.11 (m, 1H, H1); 4.45 (ddd, 1H, H3); 13C-NMR (CDCl3) δ 23.64; 24.01; 24.90; 34.84; 35.82; 38.22; 39.20 (cyclohex.; C4, C6); 60.55 (C7); 69.50; 74.47; 80.80 (C1, C2, C3); 73.92 (C5); 110.04 (OCO).
(1R,2S,3R,5R)-1,2-O-cyclohexylidene-5-C-aminomethyl-cyclohexane-1,2,3,5-tetrol (16). The azide 15 (0.85 g, 3 mmol), dissolved in 10 mL of ethanol, was hydrogenated for 4 h in the presence of Pd/C 10% (0.1 g). The catalyst was removed by filtration and the solvent was evaporated in vacuo. The residue was purified by flash chromatography on silica gel and the amine 16 was isolated in 88% yield; oil; IR (KBr) 3100-3500 (NH, OH) cm-1; 1H-NMR (C5D5N) δ 1.60 (m, 11H, cyclohex., H6; 1.88 (dd, 1H, H4, J4-4'=13.6 Hz); 2.13 (dd, 1H, H6', J6'-6= 14.6 Hz); 2.40 (m, 1H, H4'); 2.88 (2d, 2H, H7, H7', J7-7'=12.8 Hz); 4.34 (t, 1H, H2, J2-1=J2-3=6.0 Hz); 4.60 (m, 2H, H1, H3); 4.83 (sl, 4H, NH2, OH); 13C-NMR (C5D5N) δ 24.31; 24.58; 25.59; 35.40; 36.29; 38.64; 41.20 (cyclohex., C4, C6); 53.61 (C7); 68.95; 72.50; 74.40; 81.51 (C1, C2, C3, C5); 109.15 (OCO).
(1R,2S,3R,5R)-1,2-O-cyclohexylidene-3-O-(5-nitro-2-furoyl)-5-C-[(5-nitro-2-furamide)methyl]cyclo-hexane-1,2,3,5-tetrol (17). To a stirred solution of 16 (0.257 g, 1 mmol) in anhydrous CH2Cl2 at 0 °C, were slowly added 5-nitrofuroyl chloride 5 (0.53 g, 3 mmol) and pyridine (0.5 mL). The resulting mixture was stirred at room temperature for 4 h and extracted with dichloromethane. The solvent was removed by evaporation and the residue was purified by column chromatography on silica gel, to give the desired compound 17 in 60% yield; m p 129-131°C; IR (KBr) 1650 (NHC=O), 1740 (OC=O) cm-1; 1H-NMR (CDCl3) δ 1.60 (m, 12H, cyclohex., H6, H6'); 1.94 (dd, 1H, H4, J4-4'=15.4 Hz); 2.35 (dt, 1H, H4'); 3.50 (dd, 1H, H7, J7-7'=13.6 Hz; J7-NH=7.1 Hz); 3.65 (dd, 1H, H7', J7'-NH=5.7 Hz); 4.24 (t, 1H, H2, J2-1=J2-3=5.5 Hz); 4.54 (m, 1H, H1); 5.56 (m, 1H, H3); 7.15-7.40 (m, 5H, NH, Hfuranic); 13C-NMR (CDCl3) δ 23.58; 23.92; 24.83; 33.85; 34.77; 36.85; 37.83 (cyclohex., C4, C6); 48.89 (C7): 72.12; 73.82; 73.91; 76.23 (C1, C2, C3, C5); 110.79 (OCO); 111.56; 112.36; 116.26; 119.30 (C3a, C3b, C4a, C4b); 144.61; 147.71 (C2a, C2b); 151.40; 153.00 (C5a, C5b); 156.15; 156.80 (NHC=O, OC=O).

{kind=link}
{kind=link}
{kind=link}
{kind=link}