Results and Discussion
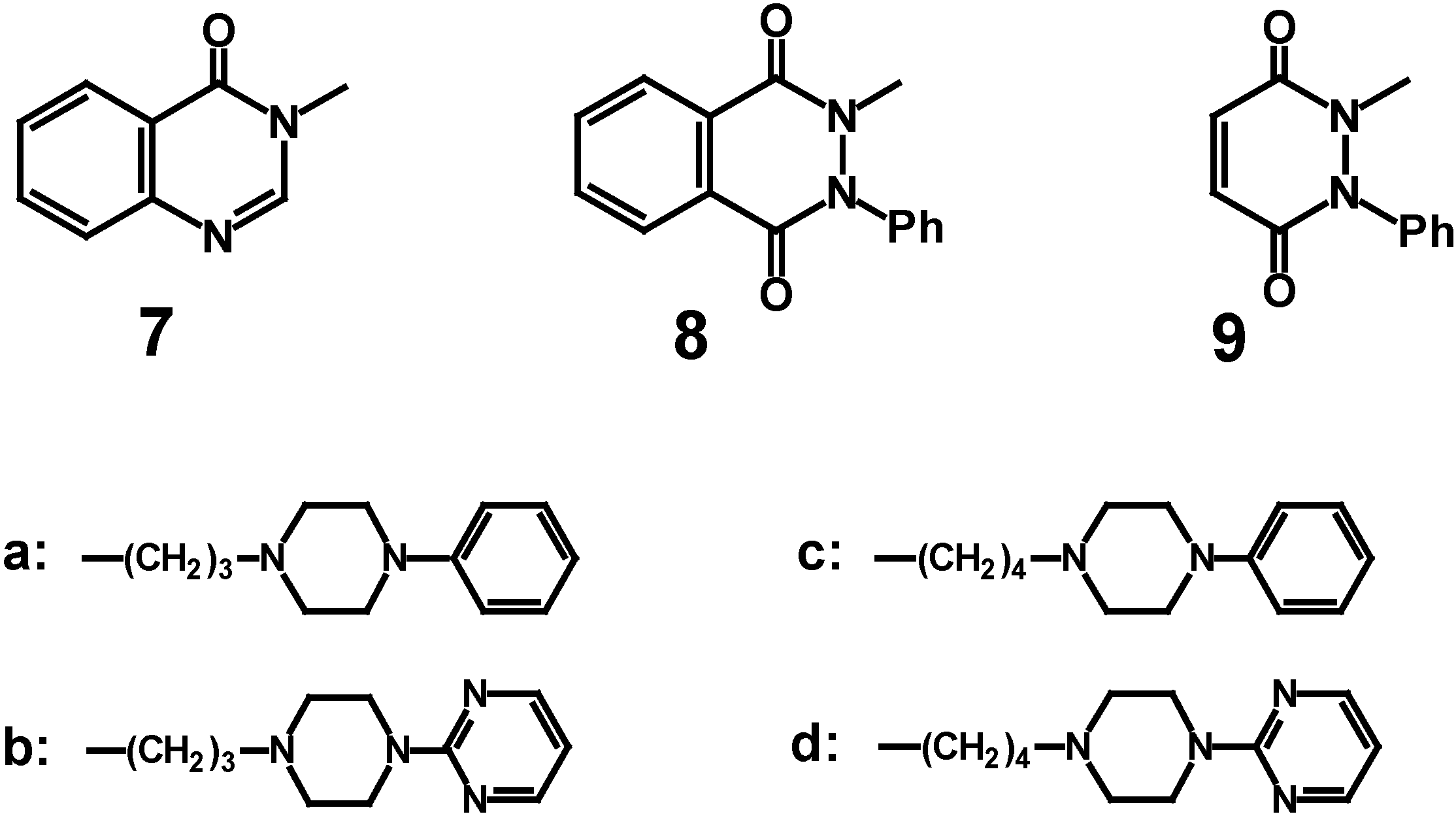
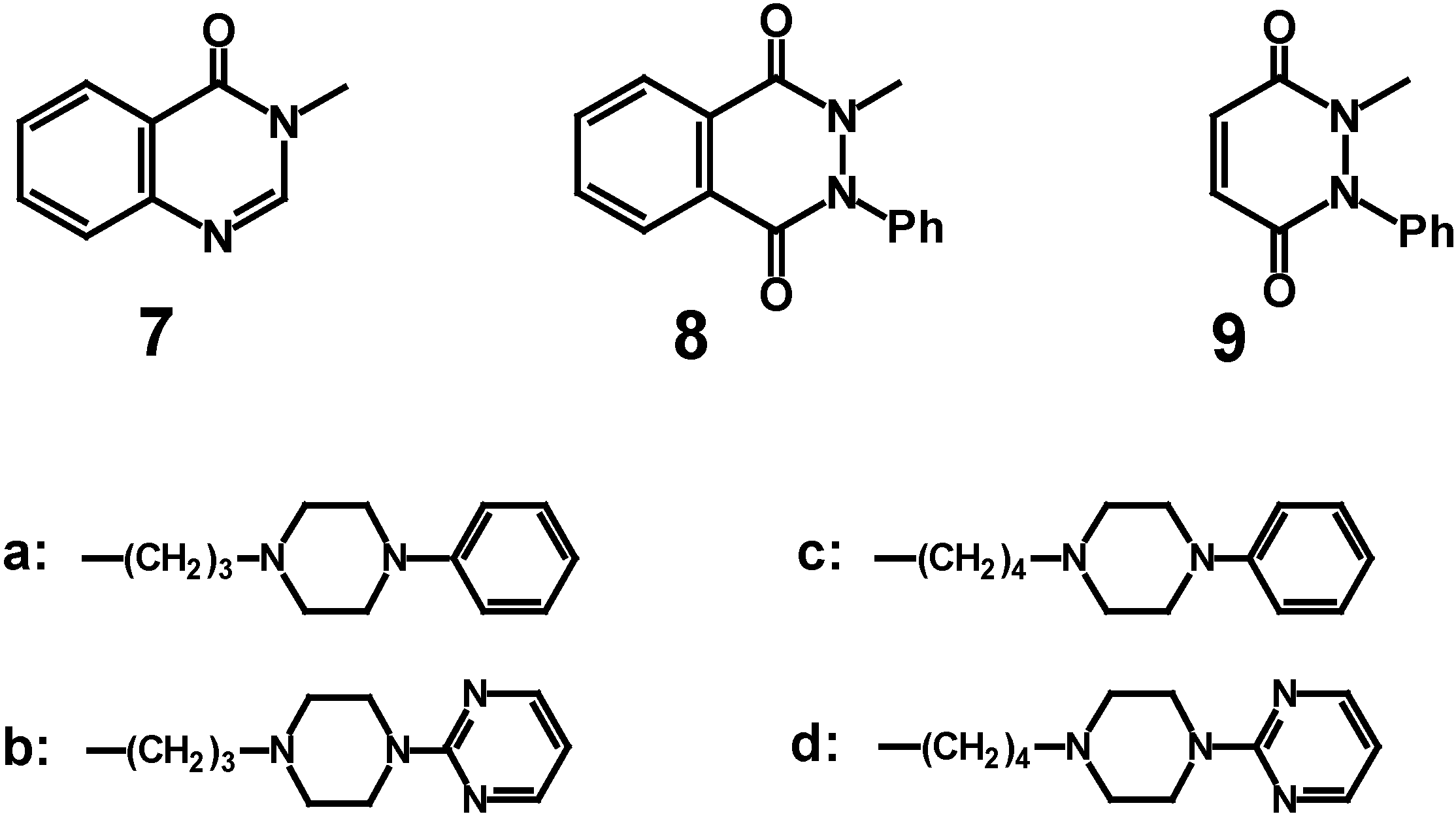
We present in this paper the synthesis of new N-[3-(4-aryl-1-piperazinyl)propyl] or N-[4-(4-aryl-1-piperazinyl)butyl] derivatives of quinazolidin-4(
3H)-one (
7a-
d), 2-phenyl-2,3-dihydrophthalazine-1,4-dione (
8a-
d) and 1-phenyl-1,2-dihydropyridazine-3,6-dione (
9a-
d) (
Scheme 1) in which the length of a spacer, arylpiperazine and terminal amide fragment were systematically modified. Preliminary investigation results on affinities of obtained compounds for 5HT
1A and 5HT
2A receptor sites are also presented.
Starting materials quinazolidin-4(
3H)-one (
7), 2-phenyl-2,3-dihydrophtalazine-1,4-dione (
8) and 1-phenyl-1,2-dihydropyridazine-3,6-dione (
9) were obtained according to procedures described in the literature. Quinazolidin-4(
3H)-one (
7) was obtained from antranilic acid [
11], 2-phenyl-2,3-dihydrophtalazine-1,4-dione (
8) from ftalic anhydride [
12], while 1-phenyl-1,2-dihydropyridazine-3,6-dione (
9) from maleic anhydride [
13].
Compounds
7a-d,
8a-
d,
9a-
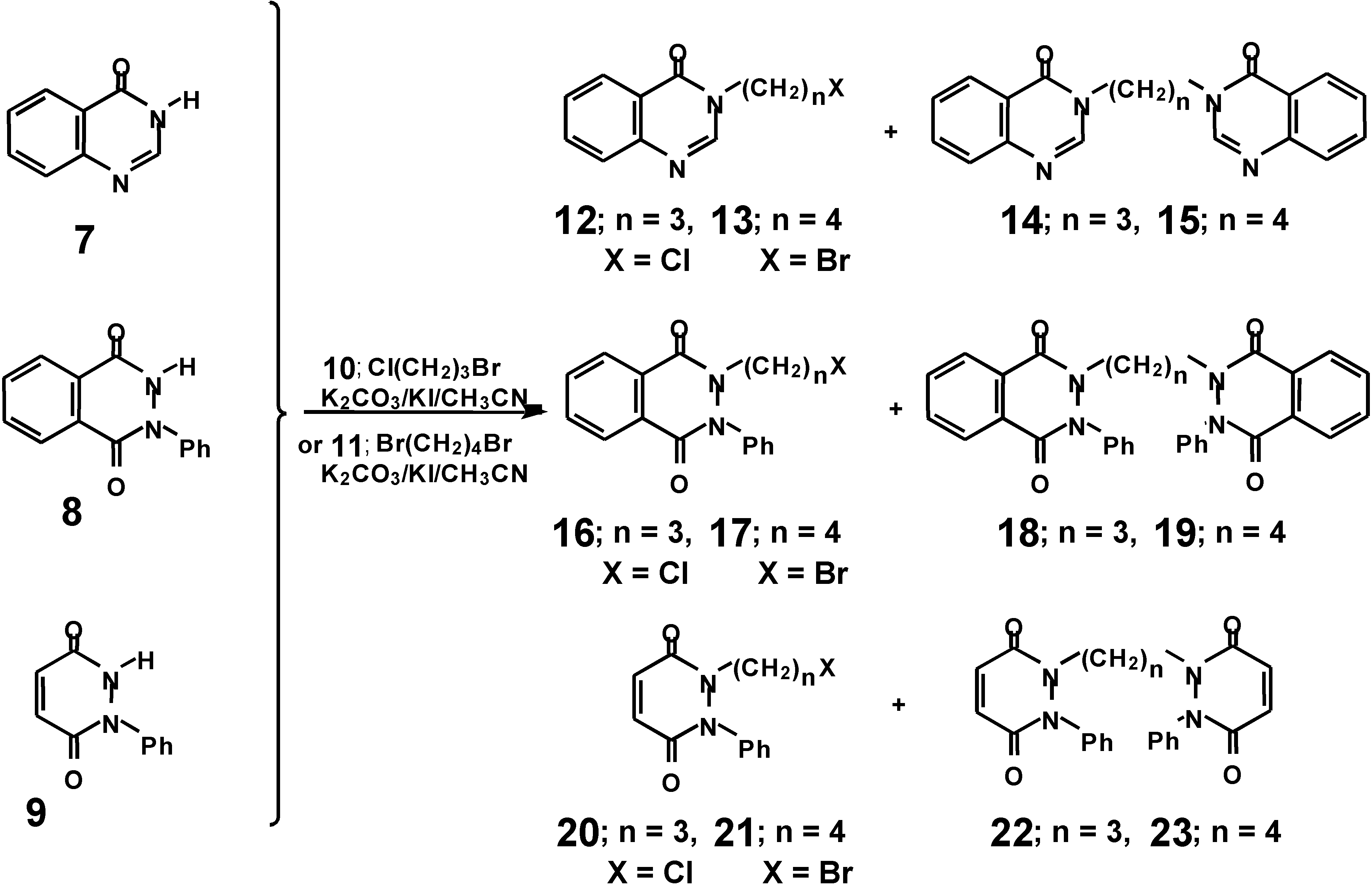
d were prepared by a two-step procedure. Alkylation of
7-
9 with 1-bromo-3-chloropropane (
10) or 1,4-dibromobutane (
11) in the presence of K
2CO
3 in acetonitrile led to the formation of halogen intermediates
12,
13,
16,
17,
20 and
21 (
Scheme 2). In the reaction, symmetrically disubstituted derivatives
14,
15,
18,
19,
22 and
23, were also formed as byproducts. When in a place of 1-bromo-3-chloropropane (
10) 1,3-dibromopropane was used, increased yields of the disubstituted derivatives (
14,
18 and
22) have been observed. The yields, melting points, and
1H-NMR signals observed in the regions characteristic of CH
2X and CH
2NC=O protons as well as absorption of the carbonyl group in IR spectra of obtained compounds
12-
23 are collected in
Table 1.
The presence of the carbonyl group in the compounds
12-
15 is confirmed by the absorptions found in the usual carbonyl region at 1658-1680 cm
-1. The
1H-NMR spectra of compounds
12-
13 display typical signals arising from the methylene hydrogens in the -CH
2X and -CH
2NC=O fragments, while disubstituted derivatives
14 and
15 gave
1H-NMR spectra in which two equivalent methylene hydrogens have been detected in the -CH
2NC=O fragment. The above analysis of the
1H-NMR and IR spectra indicated that under the applied reaction conditions quinazolidin-4(
3H)-one (
7) undergoes N-substitution. Application of the same reaction conditions to alkylation of 2-phenyl-2,3-dihydrophthalazine-1,4-dione (
8) and 1-phenyl-1,2-dihydropyridazine-3,6-dione (
9) results in formation of the N-substituted derivatives
16-
19 and
20-
23 respectively, in agreement with both our own and literature reports on the structural determination of substituted lactams [
14,
15,
16,
17,
18,
19,
20,
21].
Table 1.
Reaction yields, physical properties and spectral data of halogen- (12, 13, 16, 17, 20 and 21) and disubstituted derivatives (14, 15, 18, 19, 22 and 23) of quinazolidin-4(3H)-one (7), 2-phenyl-2,3-dihydrophthalazine-1,4-dione (8) and 1-phenyl-1,2-dihydropyridazine-3,6-dione (9).
Table 1.
Reaction yields, physical properties and spectral data of halogen- (12, 13, 16, 17, 20 and 21) and disubstituted derivatives (14, 15, 18, 19, 22 and 23) of quinazolidin-4(3H)-one (7), 2-phenyl-2,3-dihydrophthalazine-1,4-dione (8) and 1-phenyl-1,2-dihydropyridazine-3,6-dione (9).
Comp.
No. | Yield
% | M.p.
[°C] | Crystallization
solvent | 1H-NMR, δ (ppm) | IR, (cm-1)
C=O |
|---|
| CH2X | CH2NC=O |
|---|
| 12 | 51 | 105-107 | methanol | 3.61, t, 2H | 4.20, t, 2H | 1665 |
| 13 | 69 | 86-88 | methanol | 3.45, t, 2H | 4.05, t, 2H | 1658 |
| 14 | 5.3 | 194-196 | methanol | - | 4.23, t, 4H | 1672 |
| 15 | 11 | 223-225 | ethanol | - | 4.10, t, 4H | 1680 |
| 16 | 67 | 80-82 | methanol | 3.78, t, 2H | 4.52, t, 2H | 1656 |
| 17 | 49 | 65-67 | methanol | 3.52, t, 2H | 4.39, t, 2H | 1657 |
| 18 | 6.3 | 145-147 | ethanol | - | 4.61, t, 4H | 1669 |
| 19 | 28 | 241-243 | DMF | - | 4.49, t, 4H | 1657 |
| 20 | 65 | 70-72 | acetone | 3.70, t, 2H | 4.33, t, 2H | 1671 |
| 21 | 53 | 62-65 | methanol | 3.46, t, 2H | 4.20, t, 2H | 1670 |
| 22 | 7.1 | 202-204 | methanol | - | 4.33, t, 4H | 1659 |
| 23 | 13 | 165-167 | ethanol | - | 4.23, t, 4H | 1677 |
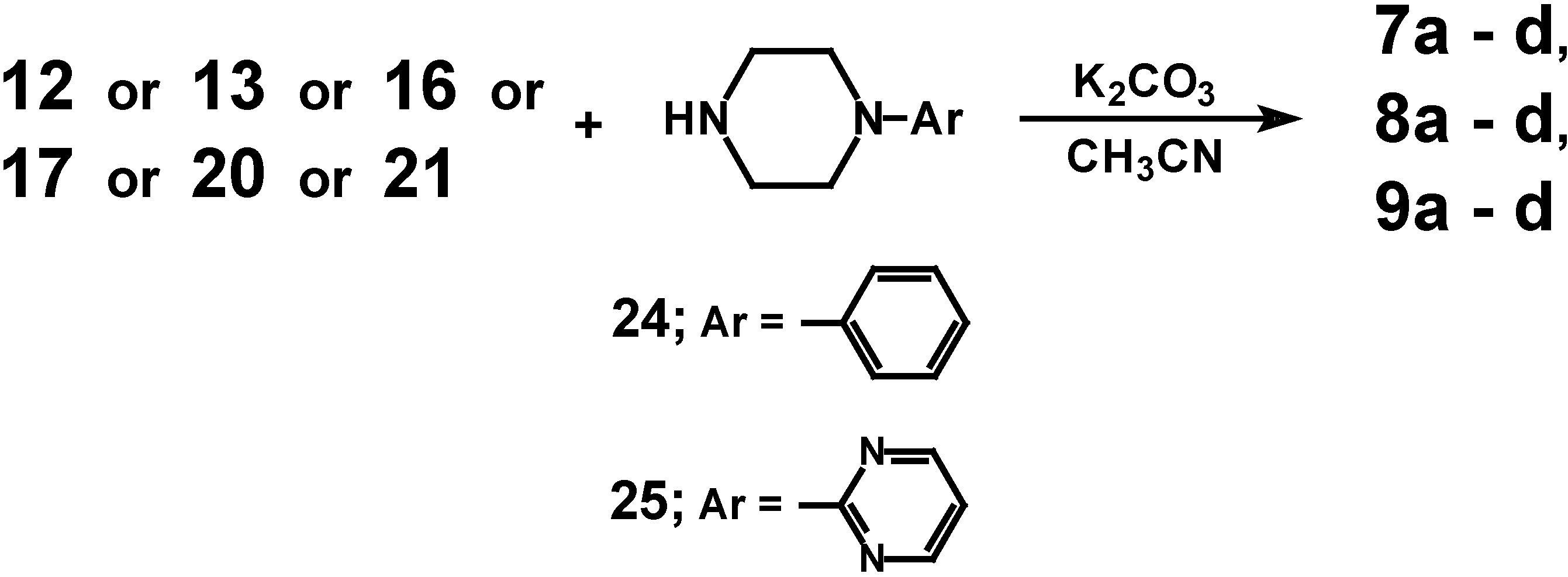
Target compounds
7a-
d,
8a-
d,
9a-
d were obtained upon condensation of intermediates
12,
13,
16,
17, 20 or
21 with 1-phenylpiperazine (
24) or 1-(2-pyrimidinyl)piperazine (
25), respectively (
Scheme 3).
Reactions were carried out in acetonitrile in the presence of anhydrous K
2CO
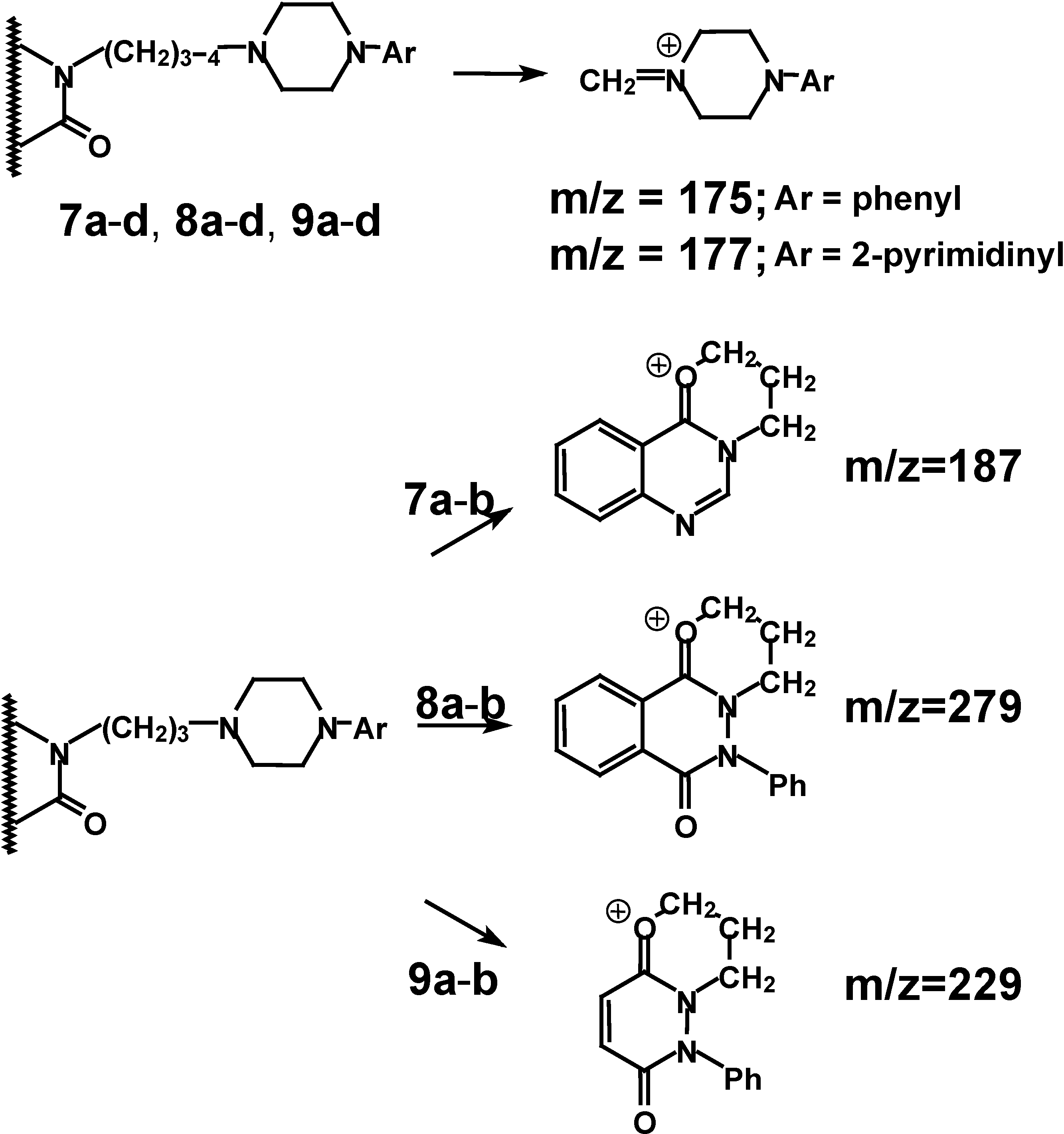
3. Reaction yields and the properties of the obtained compounds are presented in the Experimental section of the paper. The mass spectra of the compounds
7a-
d,
8a-
d,
9a-
d (
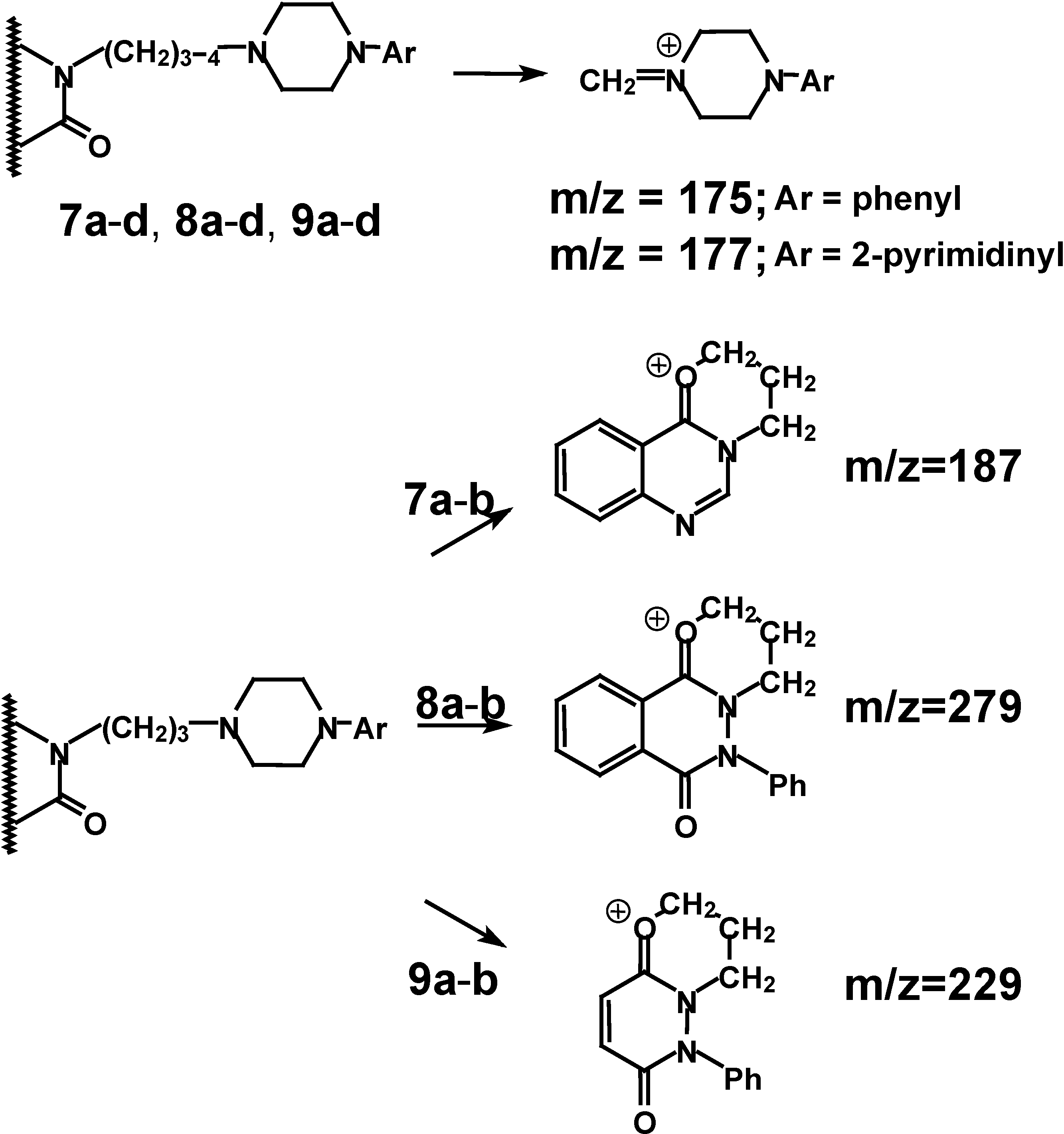
Scheme 4) generally show the presence of molecular ions of weak intensity.
The base peaks of the investigated compounds mainly correspond to [CH
2=NR
2]
+ ion formation: m/z=175; m/z=177. This is in agreement with the general patterns observed for alkylamine fragmentation [
22,
23]. In case of derivatives
7a-
b,
8a-
b,
9a-
b, with a three member spacer chain, we find that fragmentation leads to fragments with m/z=187, m/z=279 and m/z=229 respectively, which are consistent with the behaviour of alkyl substituted lactams in mass spectroscopy [
23]
Experimental
General
Elemental analyses were performed on a Perkin-Elmer 2400 analyser. EI mass spectra were carried out with a Varian MAT 112 spectrometer at 70 eV. The 1H-NMR spectra were recorded in deuterochloroform with a Tesla 487C (80 MHz) spectrometer and using tetramethylsilane (TMS) as an internal standard; the chemical shifts are reported in ppm (δ); coupling constants were taken from the expanded spectrum. IR spectra were recorded on a Bio-Rad FTS-175C spectrophotometer in KBr pellets. Melting points were determined in a Boetius apparatus and are uncorrected. For biological experiments, free bases 7a-d, 8a-d, 9a-d were converted into their hydrochloride salts and their molecular formulas and molecular weights were established on the basis of an elemental analysis.
General procedure for preparation of derivatives 12, 13, 16, 17, 20 and 21
A mixture of the lactam
7 or
8 or
9 (0.1 mole), the appropriate 1-bromo-3-chloropropane (
10) (0.12 mole) or 1,4-dibromobutane (
11) (0.12 mole), powdered K
2CO
3 (20.7 g, 0.15 mole) and a catalytic amount of KI in acetonitrile (200 mL) was stirred and refluxed 24 h (
Scheme 2). The cold reaction mixture was filtered and the filter cake washed with cold acetonitrile (20 mL). The combined filtrates were evaporated to dryness and the residue was purified by recrystallisation. Reaction yields and physical properties of the obtained compounds
12,
13,
16,
17,
20 and
21 are given in
Table 1. From the dry filter cake, after suspending in water, byproducts
14,
15,
18,
19,
22 and
23 were isolated with moderate yields. Reaction yields and physical properties of compounds
14,
15,
18,
19,
22 and
23 are collected in
Table 1.
General procedure for the preparation of compounds 7a-d, 8a-d and 9a-d
A mixture of the corresponding chloro derivative (
12,
16,
20) (0.01 mole), arylpiperazine (
24,
25) (0.01 mole), powdered K
2CO
3 (4.14 g, 0.03 mole) and catalytic amount of KI in acetonitrile (30 mL) was stirred for 48 h at 50-60° (
Scheme 3). When in place of the chloro derivatives (
12,
16,
20) a bromo derivative (
13,
17,
21) (0.01 mole) was used, the reaction mixture was stirred at 50-60 ° for 24 h. The inorganic precipitate was filtered off, the filtrate was evaporated, and the residue was recrystallised from the appropriate solvent.
General procedure for the preparation of hydrochlorides
Free bases 7a-d, 8a-d and 9a-d were converted into their hydrochlorides by dissolving the corresponding base in acetone (10mL/g) and treating with ethanol saturated with HCl. The precipitate was filtered off and washed with acetone. Some of the hydrochlorides were additionally purified by recrystallisation.
3-[3-(4-phenyl-1-piperazinyl)propyl]-quinazolidin-4(3H)-one (7a)
Base 7a was obtained in 64% yield, m.p. 119-121°C (methanol); 1H-NMR: δ 1.92-2.17 (m, 2H, CH2CH2CH2), δ 2.35-2.66 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.10-3.24 (m, 4H, (CH2)2NAr), δ 4.12 (t, 2H, CH2NC=O, J=6.6 Hz), δ 8.14 (s, 1H, CH=N), δ 6.87-8.38 (m, 9HAr); MS: m/z (I%); M 348 (92), 187 (100) 175 (56); Hydrochloride m.p. 210-213°C (acetone-methanol 10:1); Anal. Calcd. for C21H24N4O•2HCl•1.5H2O (448.40): C, 56.25; H, 6.52; N, 12.50; Found: C, 55.96; H, 6.37; N, 12.58.
3-{3-[4-(2-pyrimidinyl)-1-piperazinyl]propyl}-quinazolidin-4(3H)-one (7b)
Base 7b was obtained in 71% yield, m.p. 102-103°C (acetone); 1H-NMR: δ 1.94-2.16 (m, 2H, CH2CH2CH2), δ 2.34-2.59 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.72-3.91 (m, 4H, (CH2)2NAr), δ 4.14 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.48 (t, 1H, 5HPyrim, J=4.7 Hz), δ 8.16 (s, 1H, CH=N), δ 8.31 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz), δ 7.44-8.38 (m, 4HAr); MS: m/z (I%); M 350 (5),187 (100), 177 (30); Hydrochloride m.p. 224-227°C (acetone-ethanol 10:1); Anal. Calcd. for C19H22N6O•2HCl (423.35): C, 53.91; H, 5.71; N, 19.85; Found: C, 53.99; H, 5.77; N, 19.76.
3-[4-(4-phenyl-1-piperazinyl)butyl]-quinazolidin-4(3H)-one (7c)
Base 7c was obtained in 61% yield, m.p. 139-141°C (methanol); 1H-NMR: δ 1.56-2.03 (m, 4H, CH2CH2CH2CH2), δ 2.34-2.66 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2) , δ 3.12-3.25 (m, 4H, (CH2)2NAr), δ 4.05 (t, 2H, CH2NC=O, J=6.6 Hz), δ 8.03 (s, 1H, CH=N), δ 6.81-8.37 (m, 9HAr); MS: m/z (I%); M 362 (69), 175 (100); Hydrochloride m.p. 216-219°C (acetone-ethanol 10:1); Anal. Calcd. for C22H26N4O•2HCl (435.40): C, 60.69; H, 6.48; N, 12.87; Found: C, 60.42; H, 6.58; N, 12.60.
3-{4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl}-quinazolidin-4(3H)-one (7d)
Base 7d was obtained in 70% yield, m.p. 95-97°C (acetone); 1H-NMR: δ 1.62-2.01 (m, 4H, CH2CH2CH2CH2), δ 2.34-2.54 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.74-3.96 (m, 4H, (CH2)2NAr), δ 4.05 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.47 (t, 1H, 5HPyrim, J=4.7 Hz), δ 8.04 (s, 1H, CH=N), δ 8.30 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz), δ 7.50-8.37 (m, 4HAr); MS: m/z (I%); M 364 (21), 177 (100); Hydrochloride: m.p. 192-195°C (2-propanol-acetone 1:1); Anal. Calcd. for C20H24N6O•HCl•0.5H2O (409.92): C, 58.60; H, 6.39; N, 20.50; Found: C, 58.89; H, 6.64; N, 20.25.
3-[3-(4-phenyl-1-piperazinyl)propyl]-2-phenyl-2,3-dihydrophthalazine-1,4-dione (8a)
Base 8a was obtained in 69% yield, m.p. 54-56°C (methanol); 1H-NMR: δ 1.99-2.26 (m, 2H, CH2CH2CH2), δ 2.56-2.75 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.11-3.28 (m, 4H, (CH2)2NAr), δ 4.45 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.91-8.55 (m, 14HAr); MS: m/z (I%); M 440 (10), 279 (6), 175 (100); Hydrochloride m.p. 215-218 °C (ethanol); Anal. Calcd. for C27H28N4O2•HCl•0.5H2O (486.02): C, 66.73; H, 6.22; N, 11.53; Found: C, 66.62; H, 6.19; N, 11.50.
3-{3-[4-(2-pyrimidinyl)-1-piperazinyl]propyl}-2-phenyl-2,3-dihydrophthalazine-1,4-dione (8b)
Base 8b was obtained in 63% yield, m.p. 128-130°C (acetone); 1H-NMR: δ 1.97-2.27 (m, 2H, CH2CH2CH2), δ 2.45-2.72 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.78-3.93 (m, 4H, (CH2)2NAr), δ 4.44 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.48 (t, 1H, 5HPyrim, J=4.7 Hz), δ 8.30 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz), δ 7.34-8.55 (m, 9HAr); MS: m/z (I%); M 442 (16), 279 (56), 177 (100); Hydrochloride m.p. 226-229°C (acetone-ethanol 1:3); Anal. Calcd. for C25H26N6O2•2HCl•0.5H2O (524.45): C, 57.26; H, 5.57; N, 16.02; Found: C, 57.34; H, 5.62; N, 15.92.
3-[4-(4-phenyl-1-piperazinyl)butyl]-2-phenyl-2,3-dihydrophthalazine-1,4-dione (8c)
Base 8c was obtained in 73% yield, m.p. 111-113 °C (ethanol); 1H-NMR: δ 1.72-2.03 (m, 4H, CH2CH2CH2CH2), δ 2.41-2.70 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.14-3.29 (m, 4H, (CH2)2NAr), δ 4.39 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.86-8.47 (m, 14HAr); MS: m/z (I%); M 454 (7), 175 (100); Hydrochloride m.p. 179-183°C (acetone-ethanol 10:1); Anal. Calcd. for C28H30N4O2•HCl•H2O (491.03): C, 66.07; H, 6.13; N, 11.00; Found: C, 66.29; H, 6.50; N, 10.83.
3-{4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl}-2-phenyl-2,3-dihydrophthalazine-1,4-dione (8d)
Base 8d was obtained in 58% yield, m.p. 154-156°C (methanol); 1H-NMR: δ 1.72-1.99 (m, 4H, CH2CH2CH2CH2), δ 2.41-2.65 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.77-3.91 (m, 4H, (CH2)2NAr), δ 4.39 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.48 (t, 1H, 5HPyrim, J=4.7 Hz), δ 8.32 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz), δ 7.34-8.55 (m, 9HAr); MS: m/z (I%); M 456 (5), 177 (100); Hydrochloride m.p. 207-209°C (acetone-ethanol 10:1); Anal. Calcd. for C26H28N6O2•2HCl•H2O (547.48): C, 57.04; H, 5.89; N, 15.35; Found: C, 57.31; H, 5.98; N, 15.26.
2-[3-(4-phenyl-1-piperazinyl)propyl]-1-phenyl-1,2-dihydropyridazine-3,6-dione (9a)
Base 9a was obtained in 74% yield, m.p. 97-99°C (acetone); 1H-NMR: δ 1.88-2.09 (m, 2H, CH2CH2CH2), δ 2.44-2.69 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.12-3.26 (m, 4H, (CH2)2NAr), δ 4.25 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.85-7.74 (m, 12HAr); MS: m/z (I%); M 390 (22), 229 (14), 175 (100); Hydrochloride m.p. 219-221°C (acetone-ethanol 10:1); Anal. Calcd. for C23H26N4O2•HCl•H2O (444.96): C, 62.08; H, 6.57; N, 12.59; Found: C, 62.13; H, 6.63; N, 12.33.
2-{3-[4-(2-pyrimidinyl)-1-piperazinyl]propyl}-1-phenyl-1,2-dihydroppyridazine-3,6-dione (9b)
Base 9b was obtained in 72% yield, m.p. 152-154°C (methanol); 1H-NMR: δ 1.84-2.06 (m, 2H, CH2CH2CH2), δ 2.43-2.62 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.75-3.93 (m, 4H, (CH2)2NAr), δ 4.25 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.48 (t, 1H, 5HPyrim, J=4.7 Hz), δ 6.99-7.74 (m, 7HAr), 8.30 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz); MS: m/z (I%); M 392 (100), 229 (64), 177 (85); Hydrochloride m.p. 219-221°C (acetone-ethanol 10:1); Anal. Calcd. for C21H24N6O2•2HCl (465.38): C, 54.20; H, 5.63; N, 18.06; Found: C, 54.27; H, 5.74; N, 17.80.
2-[4-(4-phenyl-1-piperazinyl)butyl]-1-phenyl-1,2-dihydropyridazine-3,6-dione (9c)
Base 9c was obtained in 71% yield, m.p. 91-93°C (methanol-H2O 4:1); 1H-NMR: δ 1.62-1.94 (m, 4H, CH2CH2CH2CH2), δ 2.37-2.66 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.13-3.27 (m, 4H, (CH2)2NAr), δ 4.20 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.86-7.72 (m, 12HAr); MS: m/z (I%); M 404 (9), 175 (100); Hydrochloride m.p.199-202°C (methanol); Anal. Calcd. for C24H28N4O2•HCl (440.97): C, 65.37; H, 6.63; N, 12.71; Found: C, 65.12; H, 6.35; N, 12.53.
2-{4-[4-(2-pyrimidinyl)-1-piperazinyl]butyl}-1-phenyl-1,2-dihydropyridazine-3,6-dione (9d)
Base 9d was obtained in 66% yield, m.p. 104-106 °C (acetone); 1H-NMR: δ 1.56-1.81 (m, 4H, CH2CH2CH2CH2), δ 2.44-2.58 (m, 6H, CH2N(CH2)2 and CH2N(CH2)2), δ 3.75-3.91 (m, 4H, (CH2)2NAr), δ 4.20 (t, 2H, CH2NC=O, J=6.6 Hz), δ 6.48 (t, 1H, 5HPyrim, J=4.7 Hz), δ 6.97-7.74 (m, 7HAr), δ 8.30 (d, 2H, 4HPyrim and 6HPyrim, J=4.7 Hz); MS: m/z (I%); M 406 (33), 177 (100); Hydrochloride m.p. 203-205 °C (methanol); Anal. Calcd. for C22H26N6O2•HCl•H2O (460.96): C, 57.32; H, 6.34; N, 18.23; Found: C, 57.49; H, 6.19; N, 18.19.





{kind=link}
{kind=link}
{kind=link}
{kind=link}