General
Thin-layer chromatography (TLC) was accomplished on 0.2-mm precoated plates of silica gel 60 F-254 (Merck). Visualization was made with ultraviolet light (254 and 365 nm) or with a fluorescence indicator. For preparative column chromatography, silica gel 60F 254 Merck (230-240 Mesh ASTM) was used. Melting points were determined on a Kofler melting point apparatus and are uncorrected. The specific rotation [α]D were mesured with a PERKIN ELMER 141 polarimeter. 1H-NMR spectra were recorded on a BRUKER AC 300 P (300 MHz) spectrometer, 13C-NMR spectra on a BRUKER AC 300 P (75 MHz) spectrometer. Unless stated otherwise the solvent used was CDCl3, chemical shifts are expressed in parts per million downfield from tetramethylsilane used as an internal standard and δ values refer to singlet absorptions. Data are given in the following order: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad), number of protons, coupling constants J are given in Hertz. The mass spectra (HRMS) were taken on a VARIAN MAT 311 at a ionizing potential of 70 eV in the Centre Régional de Mesures Physiques de l’Ouest (CRMPO, Rennes). Absolute ethanol was distilled over magnesium after standing overnight and stored over molecular sieves (3Å). Solvents were evaporated with a Buchi rotary evaporator. All reagents were purchased from Acros, Aldrich, Avocado and Strem and were used without purification.
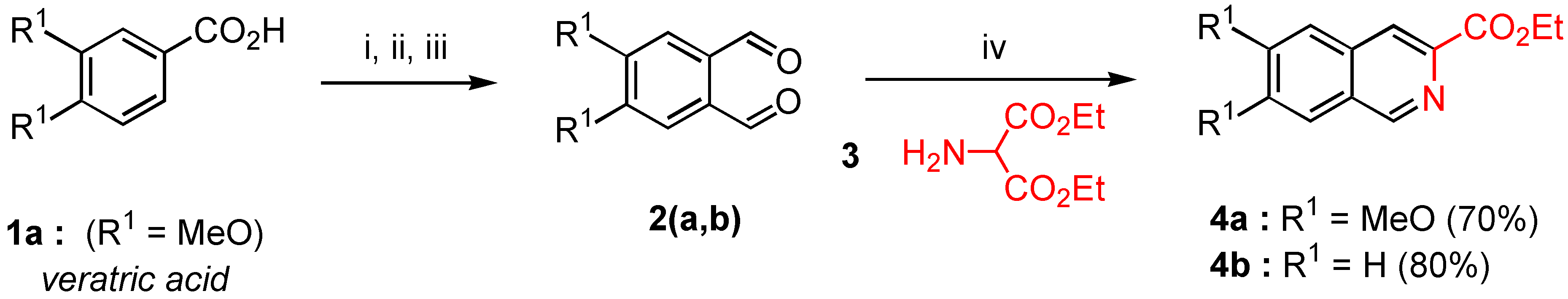
Diethyl aminomalonate (3). A solution of saturated sodium bicarbonate (225 ml) was added dropwise over 20 minutes at room temperature to a suspension of commercial diethyl aminomalonate hydrochloride (30 g., 0.14 mmol) in methylene chloride (300 mL) under vigorous magnetic stirring. After stirring for 20 minutes and decantation, the organic layer was dried over MgSO4, filtered, and the filtrate was concentrated in vacuo to give 22.6 g of the desired diethyl aminomalonate (91% yield). Compound 3 was stored under an inert atmosphere at 4°C and used without further purification. 1H-NMR (200 MHz) δ: 1.3 (t, 6H, J = 7 Hz), 2.1 (s, H), 4.25 (q, 4H, J = 7 Hz), 5.36 (s, 2H).
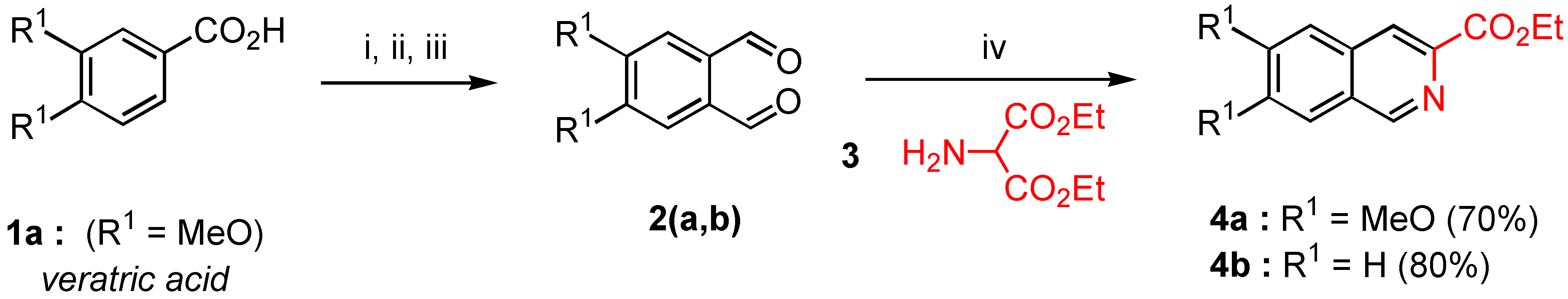
Ethyl 6,7-dimethoxy isoquinoline-3-carboxylate (4a). MgSO4 (1.2 g.) and EtONa (0.6 g ., 8.95 mmol) were successively added in one portion at 0°C under nitrogen to a solution of freshly prepared diethyl aminomalonate 3 (0.9 g., 5.15 mmol) and 4,5‑dimethoxy orthophthalaldehyde (2a, 1 g., 5.15 mmol) in anhydrous ethanol (20 mL). The reaction mixture was refluxed during 4 hours with vigorous stirring (the reaction was monitored by TLC). After elimination of solvent in a rotary evaporator, the crude reaction mixture gave an oil which crystallized on standing. Recrystallization from AcOEt gave 0.94 g. of pure compound 4a (70% yield) as yellowish needles. Mp = 174-76°C; Rf = 0.2 (AcOEt); 1H-NMR δ: 1.48 (t, 3H, J = 7.1 Hz), 4.04 (s, 3H), 4.05 (s, 3H), 4.52 (q, 2H, J = 7.1 Hz), 7.18 (s, 1H, H-5, H-8), 7.26 (s, H-8, H-5), 8.43 (s, H-4), 9.12 (s, H-1); 13C-NMR δ: 14.47 (qt, J = 127, 2.5 Hz), 52.62 (q, J = 145 Hz), 56.24 (q, J = 145 Hz), 61.64 (tq, J = 143 Hz, 4.5 Hz), 105.37 (dd, J = 160, 2.8 Hz, C-5, C-8), 105.76 (dd, J = 161, 5 Hz, C-5, C-8), 122.57 (dd, J = 165, 4.4 Hz, C-4), 126.33 (t, J = 6 Hz, C-4a, C-8a), 132.09 (t, J = 5.9 Hz, C-8a, C-4a), 140.68 (d, J = 12 Hz, C-3), 149.95 (dd, J = 179, 5 Hz, C-1), 152.03 (dm, J = 4 Hz, C-6, C-7), 153,4 (dm, J = 4 Hz, C-6, C-7), 166.03 (m, CO) ; HRMS (m/z): found 261.0998 (calc. for C14H15NO4, M+ requires: 261.1001).
Ethyl isoquinoline-3-carboxylate (4b). Crude product 4b was prepared according to the method used for the synthesis of 4a from an equimolecular mixture of commercial orthophthaladehyde (2b, 1.0 g., 7.46 mmol) and freshly prepared diethyl aminomalonate (3, 1.3 g., 7.46 mmol) with the same reaction time. After removal of the solvent in vacuo, the crude reaction mixture gave an oil which was purified by distillation under reduced pressure (with a Büchi microdistillator). 4b was obtained in 80% yield (1.2 g.) as a mobile and colourless oil (bp = 80°C / 0.4 Torr); 1H-NMR δ: 1.49 (t, 3H, J = 7.1 Hz), 4.53 (q, 2H, J = 7.1 Hz), 7.76 (m, 2H, H-6, H-7), 7.95 (d, J = 7.7 Hz, H-5, H-8), 8.04 (d, 1H, J = 7.6 Hz, H-8, H-5), 8.59 (s, 1H, H-4); 9.34 (s, 1H, H-1), 13C-NMR δ: 14.45 (qt, J = 127, 2.7 Hz), 61.85 (tq, J = 147, 4 Hz), 123.95 (dd, J = 166, 4 Hz, C-5, C-8), 127.69 (dt, J = 162, 6 Hz, C-6, C-7), 127.97 (dt, J = 163, 6 Hz, C-6, C-7), 129.56 (dd, J = 140, 8 Hz, C-5, C-8), 129.86 (m, C-4a or C-8a), 131.15 (dd, J = 154, 8 Hz, C-4), 135.14 (dm, J = 6 Hz, C-4a, C-8a), 141.74 (d, J = 12.3 Hz, C-3), 152.69 (dd, J = 180, 5 Hz, C-1), 166 (m, CO). HRMS (m/z): found 201.0798 (calc. for C12H11NO2, M+ requires: 201.0970).
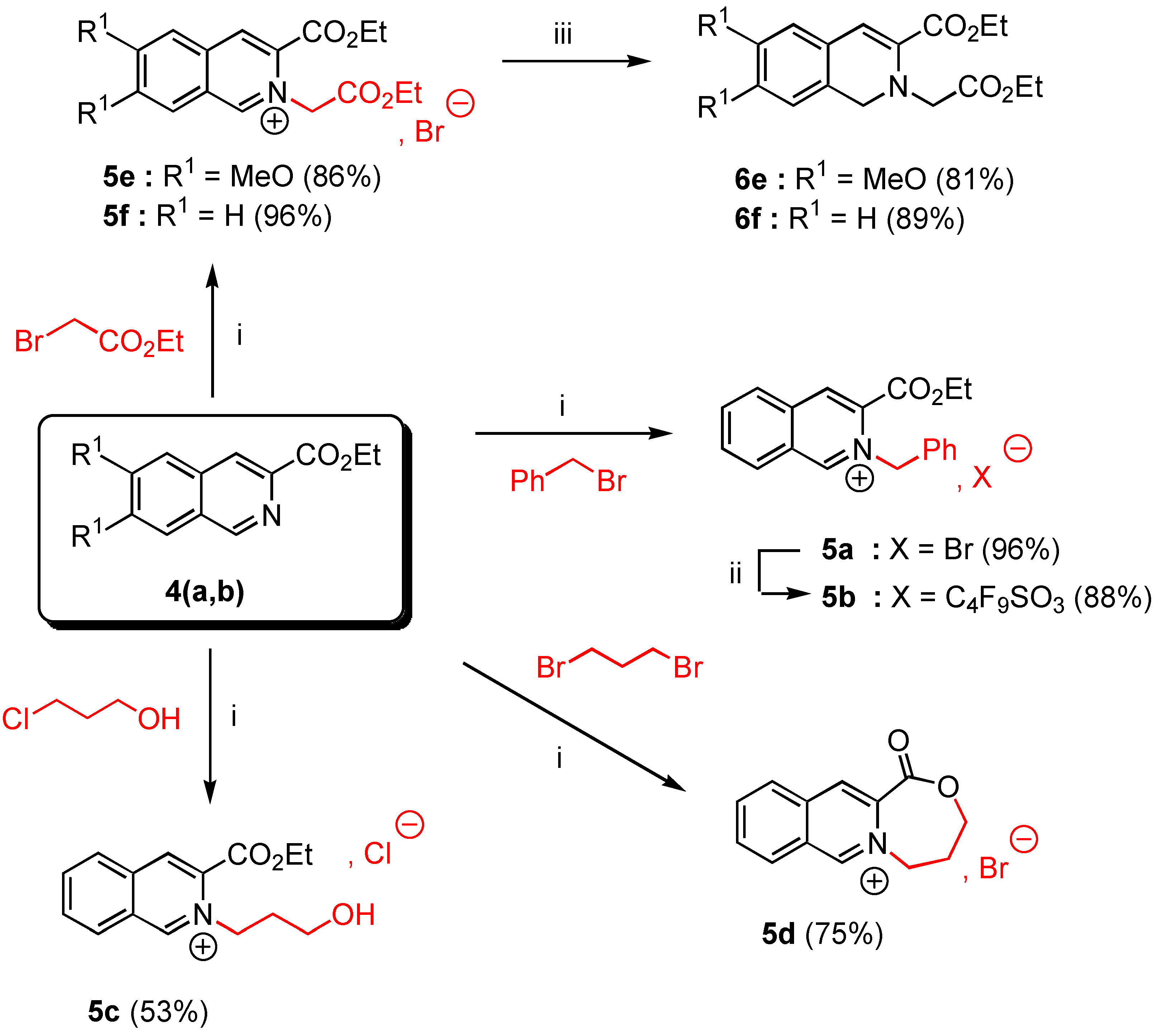
General procedure for the preparation of salts and 5a, 5c-f by the solventless N-alkylation method.
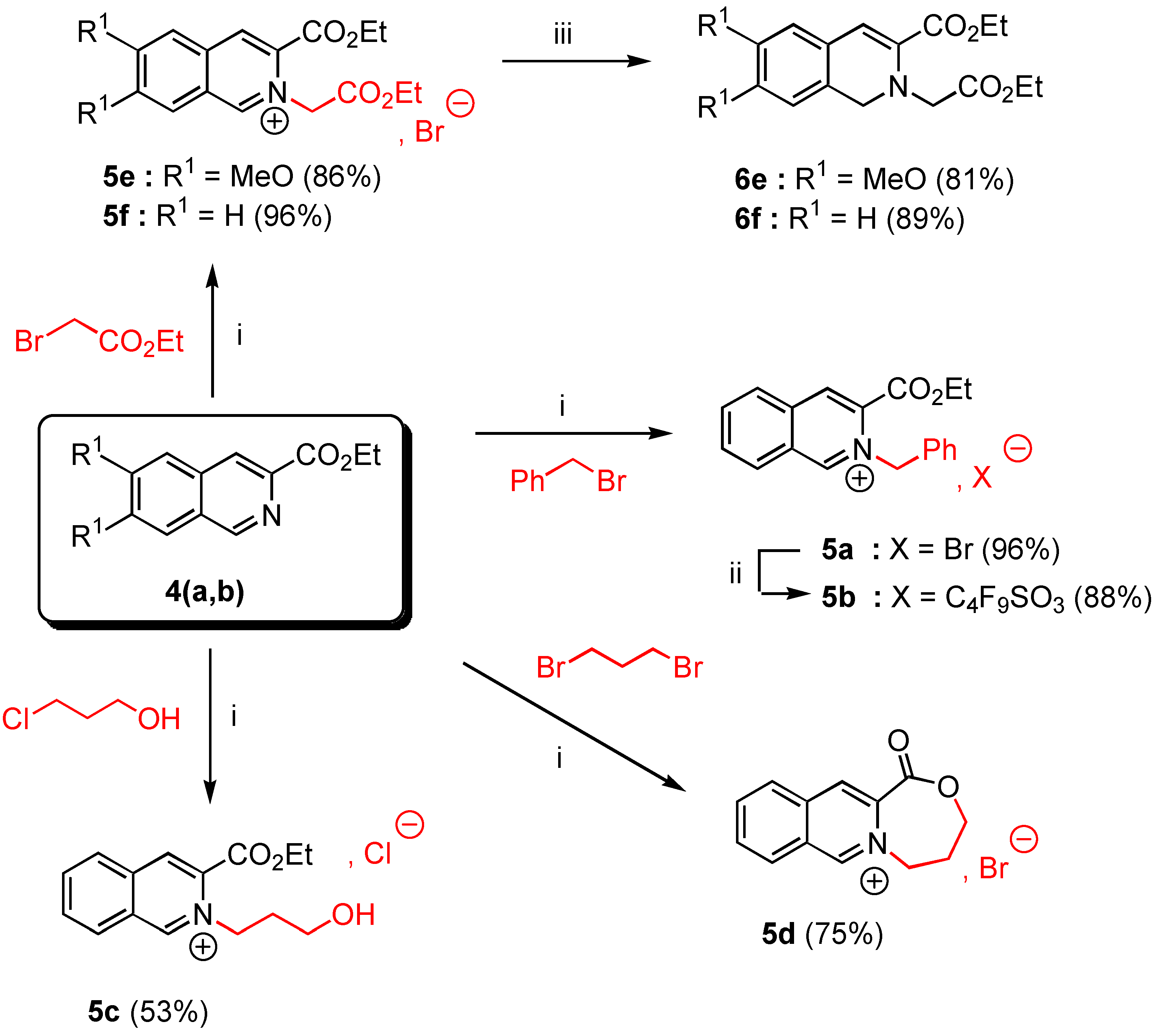
In a 50 mL two-necked flask with exclusion of moisture (CaCl2 tube) were placed 10 mmoles of ethyl 6,7-dimethoxy isoquinoline-3-carboxylate (4a, 2.61 g.) or ethyl isoquinoline-3-carboxylate (4b, 2.01 g.) and 25 mmoles of the appropriate alkyl halide (4.28 g of benzyl bromide for 5a, 2.36 g of 3‑chloropropanol for 5c, or 4.18 g of ethyl bromoacetate for 5e,f, or 5.03 g of 1,3-dibromopropane for 5d). The suspension was heated at 90°C under nitrogen during 4 hours with vigorous stirring. The reaction was allowed to cool down to room temperature and Et2O (30 mL) was added. The insoluble salt 5 was filtered off, washed twice with Et2O (20 ml) and dried over CaCl2 to give the expected salt 5 as white needles.
3-Ethoxycarbonyl-2-benzyl isoquinolinium bromide (5a). Yield = 90%; mp = 132-34°C (moisture sensitive); 1H-NMR δ: 1.32 (t, 3H, J = 7.1 Hz), 4.41 (q, 2H, J = 7.1 Hz), 6.64 (s, 2H), 7,33 (m, 5H, Ar), 8.08 (t, 1H, J = 7.6 Hz, H-6, H-7), 8.29 (t, 1H, J = 7.4 Hz, H-6, H-7), 8.46 (d, 1H, J = 8 Hz, H-5, H-8), 8.98 (d, 1H, J = 7.4 Hz, H-5, H-8), 8.99 (s, 1H, H-4), 11.82 (s, 1H, H-1); 13C‑NMR δ: 14.05 (qt, J = 128, 2.4 Hz), 62.55 (tm, J = 149 Hz), 64.54 (tq, J = 149, 4.4 Hz), 128.26 (dm, J = 139 Hz), 128.46 (m), 128.56 (dd, J = 136, 4.3 Hz), 129.30 (dd, J = 162, 4.6 Hz), 129.60 (dm, J = 161 Hz, C-6, C-7), 130.44 (dm, J = 114 Hz, C-6, C-7), 132.27 (dd, J = 134, 4.5 Hz, C-5, C-8), 133.20 (m, C-4a, C-8a), 133.34 (C-4a, C-8a), 133.54 (dd, J = 167, 7.9 Hz, C-5, C-8), 136.76 (m, C-3), 138.70 (dd, J = 165, 8 Hz, C-4), 156.00 ( dm, J = 195, 5.4 Hz, C-1), 161.00 (m, CO) ; HRMS (m/z): found 292.1337 (calc. for C19H18NO2, M+ requires : 292.1338).
3-Ethoxycarbonyl-2-(3-hydroxypropyl)-isoquinolinium chloride (5c). Yield = 53%; mp = 142-44°C (moisture sensitive); 1H-NMR δ: 1.52 (t, 3H, J = 7.1 Hz), 2.26 (tm, 2H, J = 6 Hz), 3.73 (t, 2H, J = 5.2 Hz), 4.60 (q, 2H, J = 7.1 Hz), 5.48 (t, 2H, J = 6.2 Hz); 8.10 (t, 1H, J = 7.6 Hz, H-6, H-7); 8.30 (t, 1H, J = 7.4 Hz, H-6, H-7); 8.39 (d, 1H, J = 8.2 Hz, H-5, H-8); 8.99 (d, 1H, J = 8.2 Hz, H-5, H-8); 9.00 (s, 1H, H-4); 11.37 (s, 1H, H-1); 13C NMR δ : 14.10 (qt, J = 128, 2.6 Hz); 33.90 (t, J = 130 Hz); 57.40 (tm, J = 141 Hz); 58.61 (tm, J = 142 Hz); 64.29 (tq, J = 145, 4.3 Hz); 128.10 (m, C-8a); 128.31 (dm, J = 173 Hz, C-6, C-7); 130.60 (dd, J = 172, 4.3 Hz, C-5, C-8); 132.38 (dm, J = 177 Hz, C-6, C-7); 132.70 (m, C-4a); 133.41 (dd, J = 166, 7.8 Hz, C-5, C-8); 136.40 (m, C-3); 138.38 (dd, J = 165, 4.3 Hz , C-4); 155.70 (dd, J = 187, 4.7 Hz, C-1), 160.02 (m, CO). HRMS, m/z : 260.1296 found (calc. for C15H18NO3, M+requires : 260.1287).
10-Oxo-6,7,8,10-tetrahydro-9-oxo-[5a]-azonia-cyclohepta[b]naphtalene bromide (5d). Yield = 75%, mp = 244-46°C (hygroscopic salt); 1H-NMR (D2O, H2O) δ: 2.61 (tm, 2H, J = 7.1 Hz), 4.47 (t, 2H, J = 7.1 Hz), 5.07 (t, 2H, J = 7 Hz), 8.18 (t, 1H, J = 7.1 Hz, H-6, H-7), 8.33 (t, 1H, J = 7.1 Hz, H-6, H-7), 8.38 (d, 1H, J = 7.6 Hz, H-5, H-8), 8.70 (d, 1H, J = 8 Hz, H-5, H-8), 8.85 (s, 1H, H-4), 9.89 (s, 1H, H-1), 13C-NMR (D2O, H2O) δ: 30.01 (tm, J = 133 Hz), 59.10 (tt, J = 136, 4Hz), 68.77 (tt, J = 136, 3 Hz), 131.01 (m, C-4a, C-8a), 131.30 (dt, J = 152, 4.4 Hz, C-6, C-7), 133,3 (dm, J = 172, 3.5 Hz, C-6, C-7), 133.70 (dd, J = 174, 4.6 Hz, C-5, C-8), 136.30 (dd, J = 173, 6 Hz, C-5, C-8), 137.20 (m, C4a, C-8a), 139.50 (m, C-3), 141.20 (dd, J = 174, 6 Hz, C-4), 154.01 (dm, J = 180 Hz, C-1), 167.02 (m, CO); HRMS (m/z): found 214.0869 (calc. for C13H12NO2, M+ requires: 214.0868).
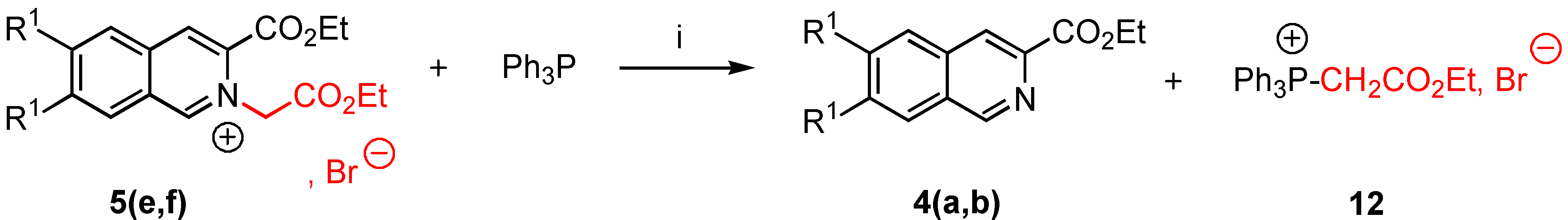
3-Ethoxycarbonyl-2-ethoxycarbonylmethyl-6,7-dimethoxy isoquinolinium bromide (5e). Yield = 86%, mp = 190-92°C (moisture sensitive); 1H-NMR δ : 1.33 (t, 3H, J = 7.1 Hz), 1.48 (t, 3H, J = 7.1 Hz), 4.13 (s, 3H), 4.24 (s, 3H), 4,27 (q, 2H, J = 7.2 Hz), 4.48 (q, 2H, J = 7.2 Hz), 6.15 (s, 2H), 7.76 (s, 1H, H-5, H-8), 8.15 (s, 1H, H-5, H-8), 8.88 (s, 1H, H-4), 11.01 (s, 1H, H-1); 13C-NMR δ : 14.06 (qt, J = 127, 2.4 Hz), 14.09 (qt, J = 127, 2.4 Hz), 57.50 (q, J = 146 Hz), 58.87 (q, J = 147 Hz), 60.10 (tm, J = 140 Hz), 62.91 (tq, J = 148, 4.7 Hz), 63.90 (tq, J = 148, 4.7 Hz), 106.98 (dm, J = 153 Hz, C-5, C-8), 109.01 (dm, J = 154 Hz, C-5, C-8), 124.95 (m, C-4a, C-8a), 127.60 (dd, J = 173, 5 Hz, C-4), 130.56 (m, C-8a, C-4a), 135.04 (m, C-3), 151.53 (dm, J = 188 Hz, C-1), 154.70 (m, C-6, C-7), 159.41 (m, C‑6, C-7), 160.01 (m, CO), 166.03 (m, CO); HRMS (m/z): found 348.1447 (calc. for C18H22NO6, M+ requires: 348.1447).
3-Ethoxycarbonyl-2-ethoxycarbonylmethyl isoquinolinium bromide (5f). Yield = 96%, mp = 152‑54°C (moisture sensitive); 1H-NMR δ: 1.33 (t, 3H, J = 7.1 Hz), 1.50 (t, 3H, J = 7.2Hz), 4.28 (q, 2H, J = 7.2 Hz), 4,52 (q, 2H, J = 7.2 Hz), 6.36 ( s, 2H, H-9), 8.10 ( t, J = 7.2 Hz, H-6, H-7), 8.30 ( t, J = 7.2 Hz, H-6, H-7), 8.43 (d, 1H, J = 7.1 Hz, H-5, H-8), 8.90 (d, 1H, J = 7.2 Hz, H-5, H-8), 9.10 (s, 1H, H-4), 11.59 (s, 1H, H-1); 13C-NMR δ: 14.16 (qt, J = 127, 2.5 Hz), 14.20 (qt, J = 128, 2.5 Hz), 61.20 (tm, J = 130 Hz), 63.47 (tq, J = 137, 4.6 Hz), 64.68 (tq, J = 139, 4.5 Hz), 128.00 (m, C-8a), 128.50 (dm, J = 152 Hz, C-6, C-7), 130.10 (dd, J = 169, 4.5 Hz, C-5, C-8), 132.30 (m, C-4a), 132.50 (dm, J = 196 Hz, C-6, C-7), 133.95 (m, C-3), 139.06 (dd, J = 158, 8 Hz, C-4), 156.90 (dm, J = 195 Hz, C-1), 160.01 (m, CO) ; 166.20 (m, CO); HRMS (m/z): found 288.1237 (calc. for C16H18NO4, M+ requires: 288.1236).
3-Ethoxycarbonyl-2-benzyl isoquinolinium 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (5b). A stirred mixture of 3-ethoxycarbonyl-2-benzyl isoquinolinium bromide (5a, 1 g., 2.68 mmol) and 2.5 equivalents of commercial potassium 1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (2.3 g., 6.72 mmol) in anhydrous EtOH (20 mL) was refluxed for 12 hours. After filtration and removal of the solvent in vacuo, the crude reaction mixture was triturated with dry Et2O (20 mL). After standing for 2 hours, the precipitated product was filtered off, washed with Et2O (2 x 20 mL) and dried under reduced pressure during 1 hour. The salt 5b was obtained in 86% yield (2.04 g.) as colourless needles, mp = 92-94°C; 1H-NMR δ: 1.32 (t, 3H, J = 7.1 Hz), 4.42 (q, 2H, J = 7.1 Hz), 6.63 (s, 2H), 7.31 (m, 5H, Ar), 8.08 (t, 1H, J = 7.6 Hz, H-6, H-7), 8.29 (t, 1H, J = 7.4 Hz, H-6, H-7), 8.46 (d, 1H, J = 8 Hz, H-5, H-8), 8.97 (d, 1H, J = 7.4 Hz, H-5, H-8), 8.98 (s, 1H, H-4), 10.90 (s, 1H, H-1); 13C-NMR δ: 14.01 (qt, J = 128, 2.4 Hz), 62.55 (tm, J = 149 Hz), 64.54 (tq, J = 149, 4.4 Hz), 128.26 (dm, J = 139 Hz), 128.46 (sm), 128.56 (dd, J = 136, 4.3 Hz), 129.30 (dd, J = 162, 4.6 Hz), 129.60 (dm, J = 161 Hz, C-6, C-7), 130.44 (dm, J = 114 Hz, C-6, C-7), 132.27 (dd, J = 134, 4.5 Hz, C-5, C-8), 133.20 (m, C-4a, C-8a), 133.34 (C-4a, C-8a), 133.54 (dd, J = 167, 7.9 Hz, C-5, C-8), 136.76 (m, C-3), 138.70 (dd, J = 166, 8 Hz, C-4), 156.01 ( dm, J = 195, 5.4 Hz, C-1), 161.02 (m, CO); HRMS (m/z): found 883.2103 (calc. for C42H36N2O7F9S, 2C+, A- requires : 883.2100).
Ethyl 2-ethoxycarbonylmethyl-6,7-dimethoxy-1,2-dihydroisoquinoline-3-carboxylate (6e). To a suspension of 3-ethoxycarbonyl-2-ethoxycarbonylmethyl-6,7-dimethoxy isoquinolinium bromide (5e, 0.5 g., 1.16 mmol) in anhydrous EtOH (5 mL) previously cooled to 0°C and vigorously stirred, was added in small portions NaBH4 (0.09 g., 2.45 mmol) under nitrogen. The mixture was then stirred at 0°C for 4 hours. TLC analysis revealed a quantitative reaction and a single product. The solvent was removed in vacuo and methylene chloride (20 mL) was added to the crude reaction mixture. After work-up (brine wash: 2 x 20 mL), the organic layer was dried over anhydrous MgSO4. Removal of the solvent by rotary evaporation gave a yellowish viscous oil which was purified by gravity chromatography on silica gel 60F-254 (Merck) using methylene chloride as eluent. Concentration of the desired fraction (Rf = 0.6) gave the expected product 6e as a nearly pure oil (81% yield). It is recommendable to handle it in the dark under an inert atmosphere at 4°C. 1H-NMR (200 MHz) δ: 1.20 (t, 3H, J = 7.2 Hz); 1.31 (t, 3H, J = 7.2 Hz); 3.83 (s, 6H); 3.98 (s, 2H); 4.11 (q, 2H, J = 7.2 Hz); 4.22 (q, 2H, J = 7.1 Hz); 4.45 (s, 2H, H-1); 6.52 (s, 1H, H-4); 6.64 (s, 1H, H-5); 6.77 (s, 1H, H-8).
Ethyl 2-ethoxycarbonylmethyl-1,2-dihydroisoquinoline-3-carboxylate (6f). The crude product 6f was synthetized according to the experimental procedure used for 6e, from 3-ethoxycarbonyl-2-ethoxy-carbonylmethyl isoquinolinium bromide (5f, 0.346 g., 1.2 mmol). 6f was purified by gravity chromatography on silica gel 60F-254 (Merck) with CH2Cl2 as eluent. Removal of the solvent in vacuo of the desired fraction (Rf = 0.4) gave pure 6f in 89% yield. Compound 6f was also stored in the dark under an inert atmosphere. 1H-NMR (200 MHz) δ: 1.23 (t, 3H, J = 7.1 Hz), 1.38 (t, 3H, J = 7.1 Hz), 4.09 (s, 2H), 4.20 (q, 2H, J = 7.1 Hz), 4.31 (q, 2H, J = 7.1 Hz), 4.44 (s, 2H, H-1), 6.83 (s, 1H, H-4), 7.15 (m, 4H, Ar).
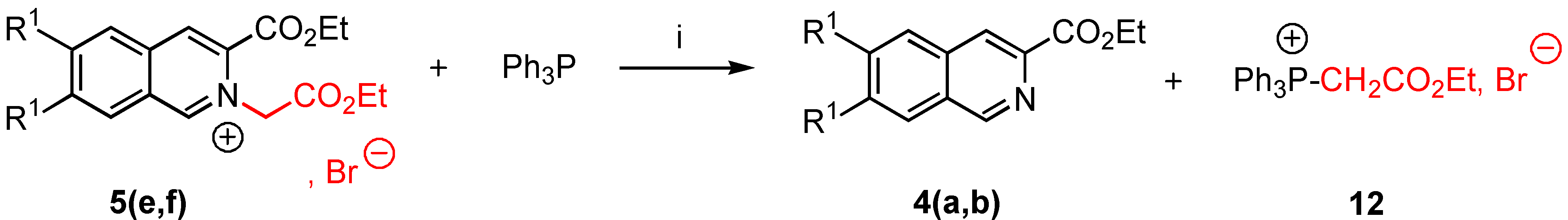
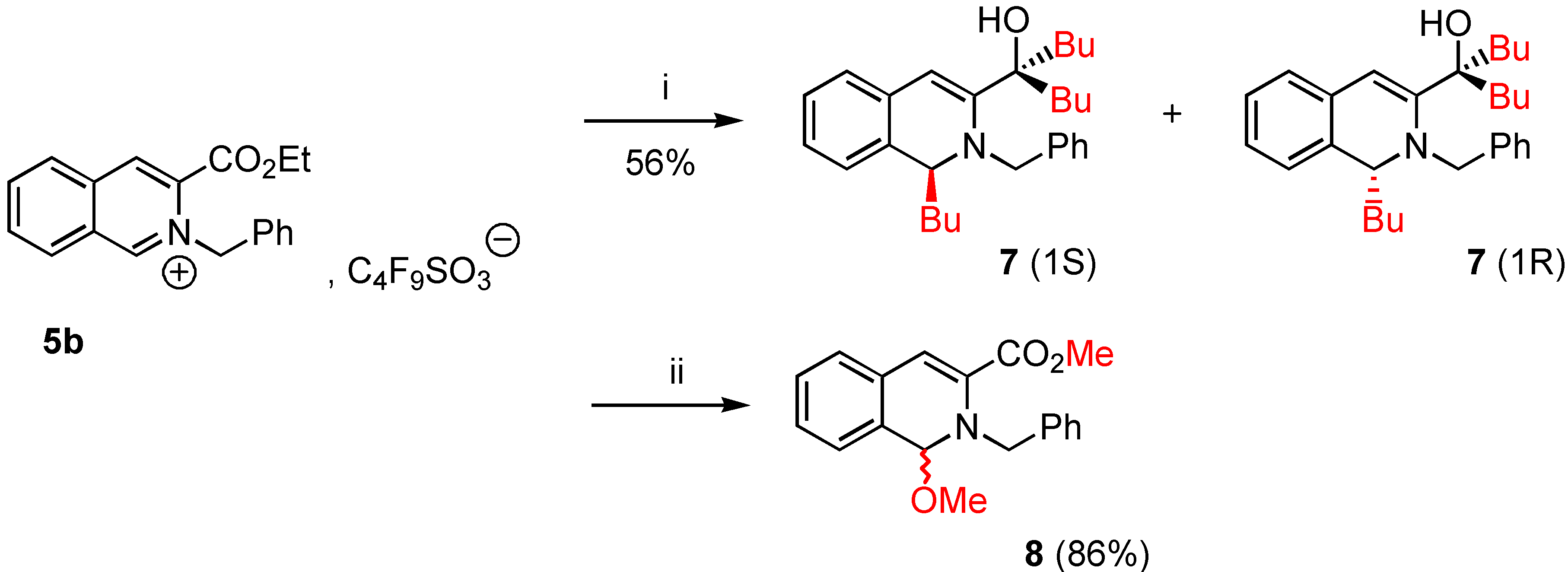
5-(1-Butyl-2-benzyl-1,2-dihydro-isoquinolin-3-yl)nonan-5-ol (7a). To a stirred suspension of 3-ethoxy-carbonyl-2-benzyl isoquinolinium 1,1,2,2,3,3,4,4,4,-nonafluorobutane-1-sulfonate (5b, 0.74 g., 0.84 mmol) in dry THF (5 mL) was added dropwise over 15 minutes at 0°C under nitrogen, a solution of nBuLi (0.2 g., 2.69 mmol, from commercial n-butyl lithium 1.6M solution in hexane) in anhydrous THF (2 mL). Stirring was continued for an additional 12 hours at room temperature. The reaction mixture was allowed to warm to 0°C, THF (10 mL) and saturated NH4Cl (20 mL) were added successively. When the mixture reached room temperature, the phases were separated and the aqueous layer was extracted twice with THF (10 mL). The combined extracts were dried over MgSO4 and the solvents removed under reduced pressure yielding a viscous oil. The crude product 7a was purified by column chromatography on silica gel 60F-254 (Merck) with 1 :1 Et2O/CH2Cl2 as eluent. The desired fraction (Rf = 0.7) was concentrated in vacuo and gave 0.2 g. of pure 7a (56% yield) as an oil [α]D24 + 1.2 (0.5, abs. EtOH). 1H-NMR δ: 0.76 (t, 3H, J = 7.2 Hz), 0.80 (t, 3H, J = 7 Hz), 0.91 (t, 3H), 1.25-1.45 (m, 12H), 1.68 (td, 2H, J = 8 Hz), 1.71-1.76 (m, 4H), 3.54 (d, 1H, Jgem = 14.1 Hz), 3.79 (dd, 1H, J = 7 Hz, H-1), 4.20 (d, 1H, Jgem = 14 Hz), 6.19 (s, 1H, H-4), 6.89-7.32 (m, 5H, Ar), 13C NMR δ: 13.94 (qt, J = 124, 4 Hz), 14.18 (qt, J = 124, 4 Hz), 15.40 (qt, J = 125, 3 Hz), 22.41, 23.08, 23.30, 25.90, 26.31, 28.30, 33.81, 40.68, 42.97, 59.10 (dt, J = 132, 4.5 Hz; C-1), 65.90 (tm, J = 134 Hz), 113.01 (dd, J = 163, 5 Hz, C-4), 124.70, 126.71, 126.90, 127.00, 127.25, 128.31, 128.42, 128.60, 131.11 (sm, C-4a, C-8a), 133.44 (m, C-4a, C-8a), 138.38 (m, C-3), 151.01 (m, CO); HRMS (m/z): found 419.3150 (calc. for C29H41NO, M+ requires: 419.3188).
Methyl 1-methoxy-2-benzyl-1,2-dihydroisoquinoline-3-carboxylate (8a). Commercial grade sodium methoxide (0.18 g., 3.39 mmol) was added in small portions under nitrogen at 0°C to a stirred suspension of 3-ethoxycarbonyl-2-benzyl-isoquinolinium-1,1,2,2,3,3,4,4,4-nonafluorobutane-1-sulfonate (5b, 1 g., 1.13 mmol) in dry methanol (3 mL). Stirring was continued for 6 hours at room temperature. The reaction mixture was evaporated under reduced pressure and methylene chloride (3 mL) was added to the residue. After elimination of compounds unsoluble in CH2Cl2, the filtrate was concentrated by rotary evaporation to give 0.3 g. of the expected compound 8a (86% yield). Compound 8a was immediately analysed by NMR. 1H-NMR δ: 3.17 (s, 3H), 3.76 (s, 3H), 4.56 (d, 1H, Ha, Hb, Jgem = 16 Hz), 5.12 (d, 1H, Hb, Ha, Jgem = 16 Hz), 5.57 (s, 1H, H-1), 6.88 (s, 1H, H-4), 7.11-7.30 (m, 9H, Ar); 13C-NMR δ: 52.12 (q, J = 147 Hz), 52.19 (q, J = 146 Hz), 56.12 (tm, J = 137, 4Hz), 89.66 (dt, J = 157, 4.7 Hz; C-1), 113.30 (dd, J = 168, 4.9 Hz, C-4), 125.73, 127.01, 127.30, 127.61, 128.22, 128.31, 128.40 (sm, C-Ar), 128.71 (sm), 130.51 (m, C-4a, C-8a), 133.14 (m, C-8a, C-4a), 139.01 (sm, C-3), 165.03 (sm, CO); HRMS (m/z): found 309.1357 (calc. for C19H19NO3, M+ requires: 309.1365).
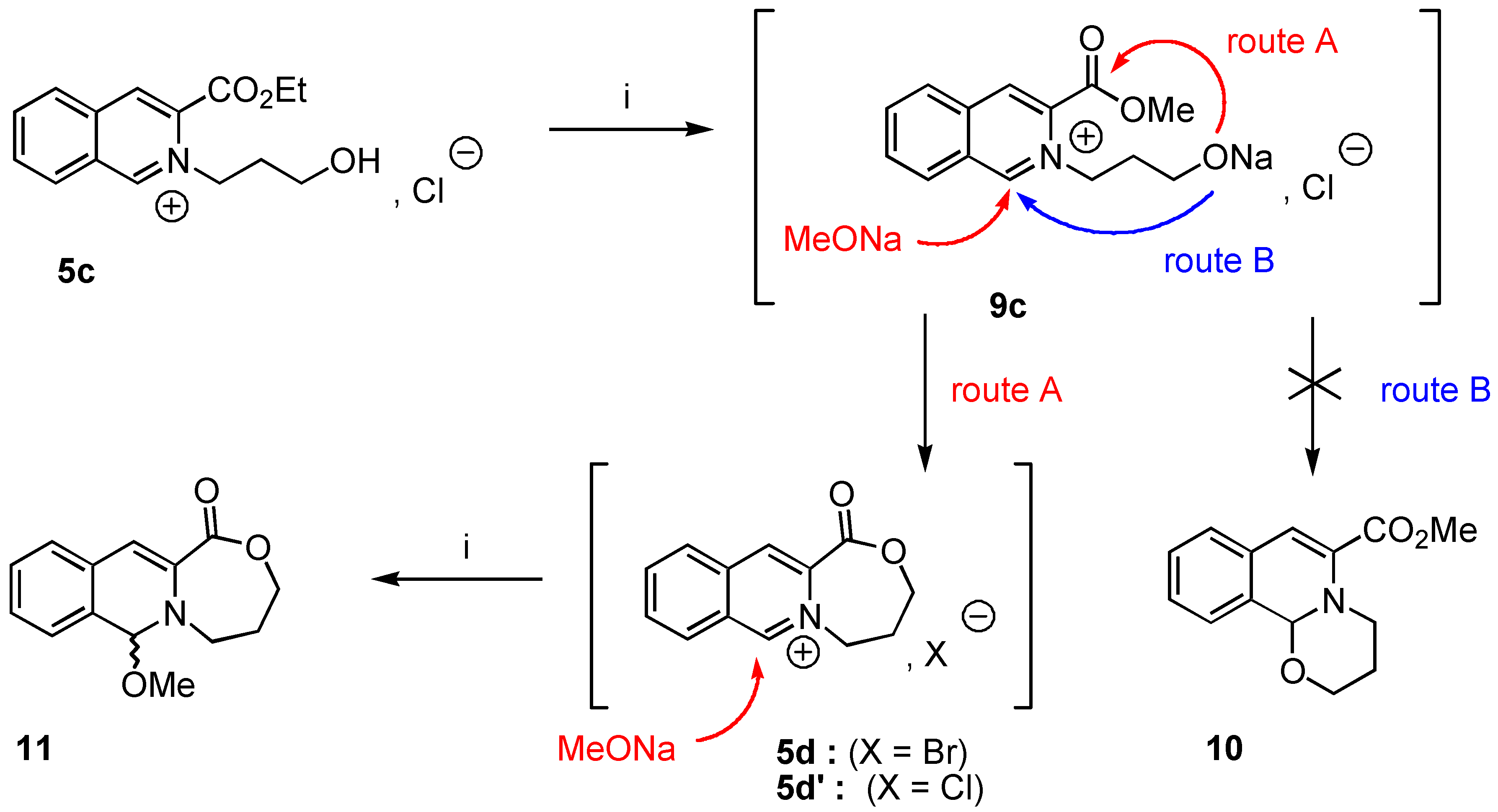
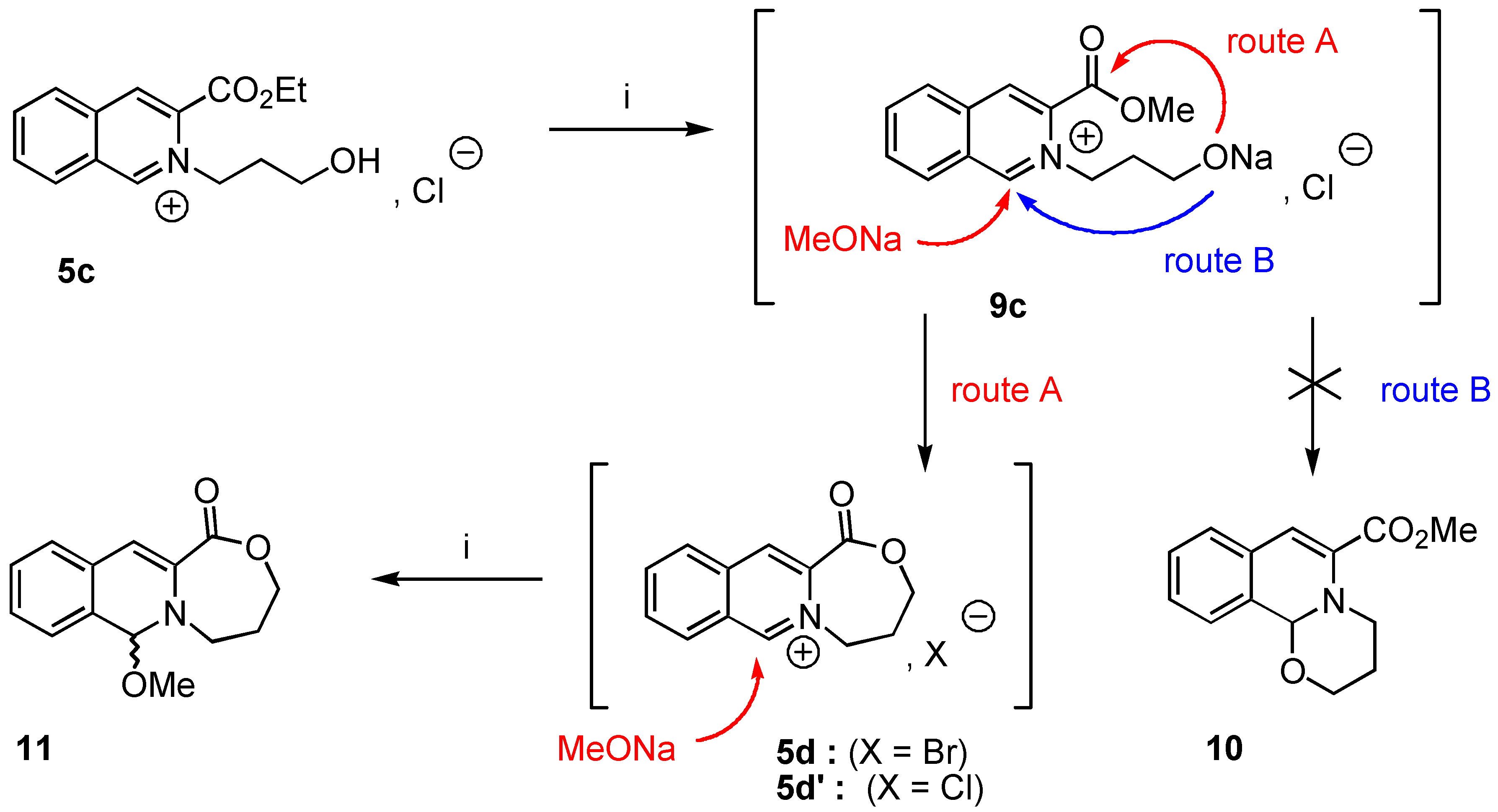
5-Methoxy-7,8-dihydro-[5H,6H]-9-oxa-[5a]-aza-cyclohepta-[b]-naphtalene-10-one (11). Crude 11 was prepared from 3-ethoxycarbonyl-2-(3-hydroxypropyl)-isoquinolinium chloride (5c, 1 g., 3.85 mmol) using the experimental procedure used for the preparation of 8a. Yield : 84% ; 1H‑NMR δ: 1.45 (dt, 1H, Jgem = 13.5 Hz, J = 1.7 Hz, Ha, Hb), 2,12 (dtm, 1H, Jgem = 13.1 Hz, J = 2 Hz, Hb, Ha), 3.37 (td, 1H, Jgem = 12 Hz, J = 2.58 Hz, He, Hf), 3.82 (s, 3H), 4.07 (td, 2H, Jgem = 12 Hz, J = 2,43 Hz, Hc, Hd), 4.67 (dm, Jgem = 12.1 Hz, J = 1.8 Hz, Hf, He), 5.89 (s, 1H, H-1), 6.54 (s, 1H, H-4), 7.11-7.30 (m, 4H, Ar); 13C-NMR δ: 27.26 (tm, J = 128 Hz), 47.72 (tm, J = 144 Hz), 52.24 (q, J = 147 Hz), 68.41 (tm, J = 147 Hz), 89.01 (dm, J = 159 Hz, C-1), 109.21 (dd, J = 163, 5.2 Hz, C-4), 125.23 (dt, J = 159, 6 Hz, C-7, C-6), 126.63 (m, C-4, C-8a), 127.47 (dt, J = 135, 4 Hz, C-6, C-7), 127.70 (dd, J = 153, 7 Hz, C-5, C-8), 129.21 (dd, J = 5Hz, C-8, C-5), 130.63 (sm, C-8, C-4a), 134.03 (sm, C-3), 165.01 (sm, CO); HRMS (m/z): found 245.1031 (calc. for C14H15NO3, M+ requires : 245.1052).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}