Total Synthesis of (-)-(7S,10R)-Calamenene and (-)-(7S,10R)-2-Hydroxycalamenene by Use of a Ring-Closing Metathesis Reaction. A Comparison of the cis- and trans-Isomers

Abstract
:Introduction
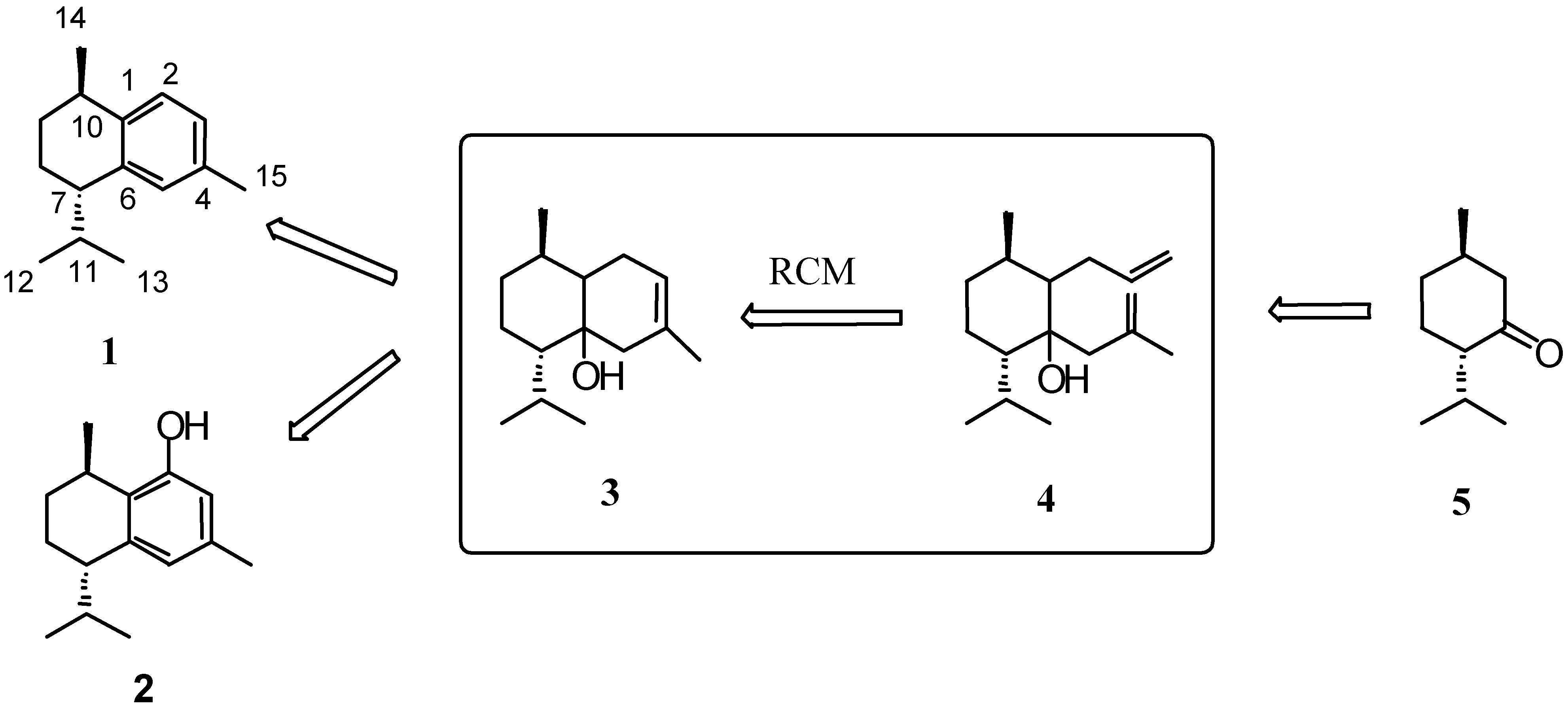
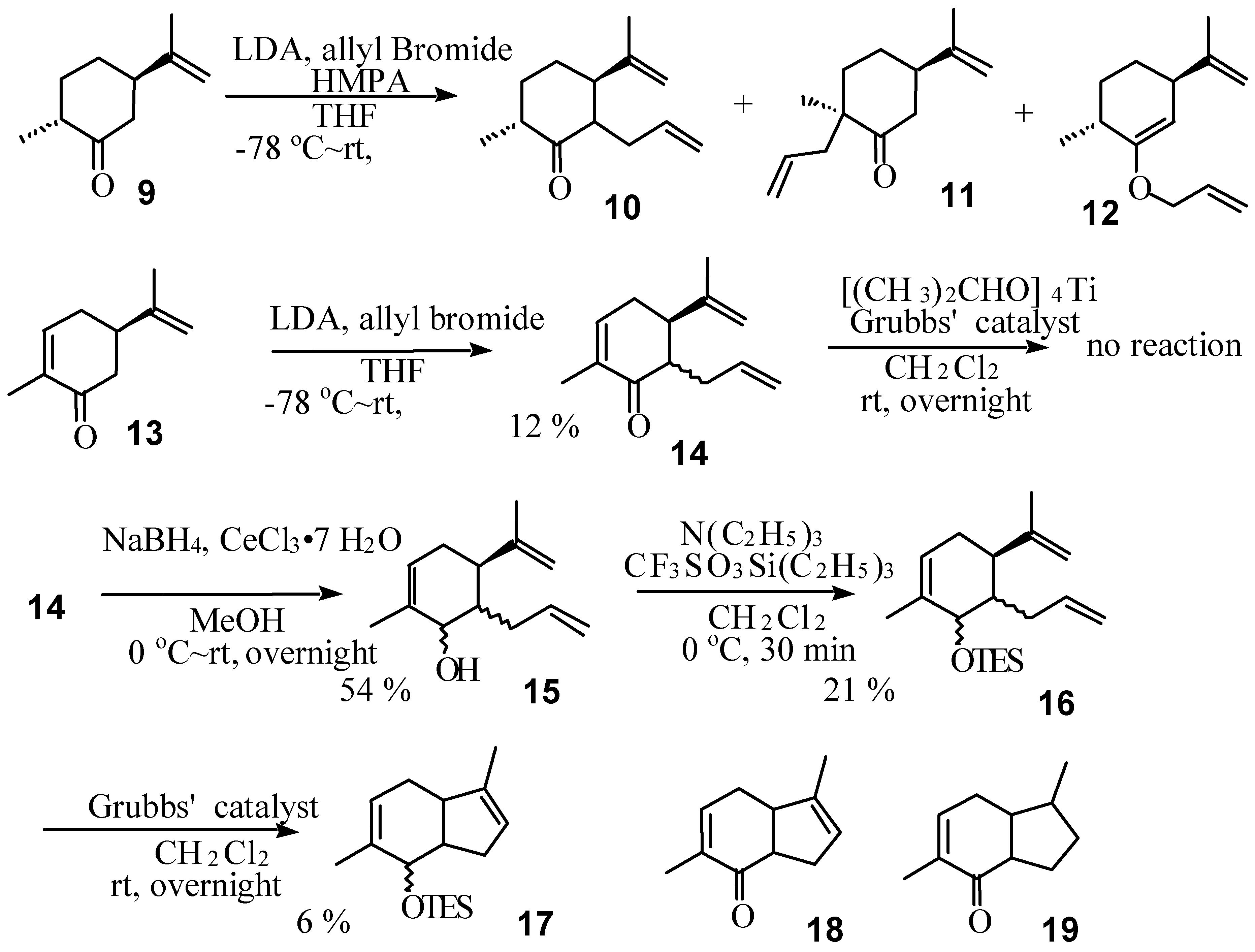
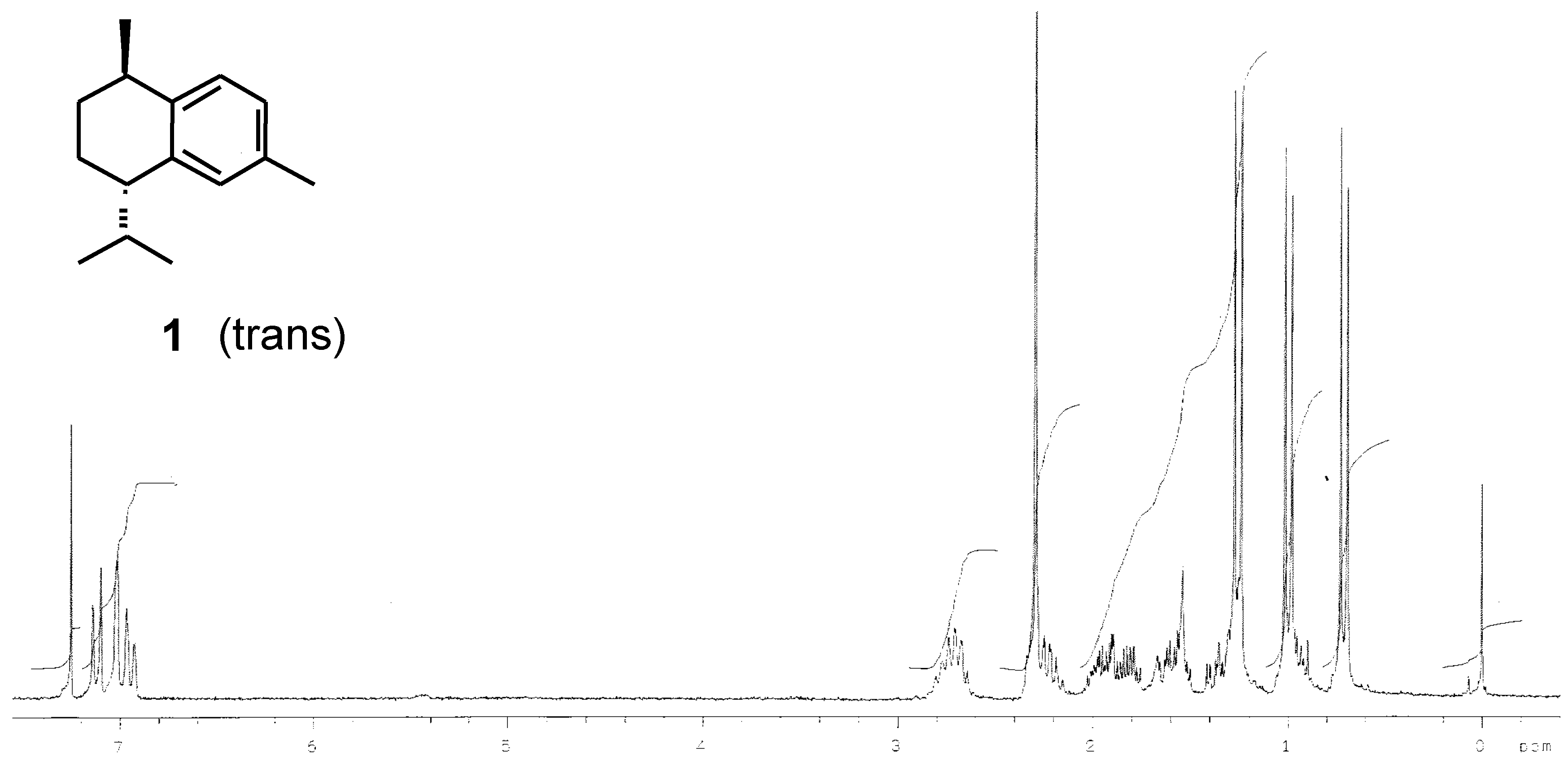
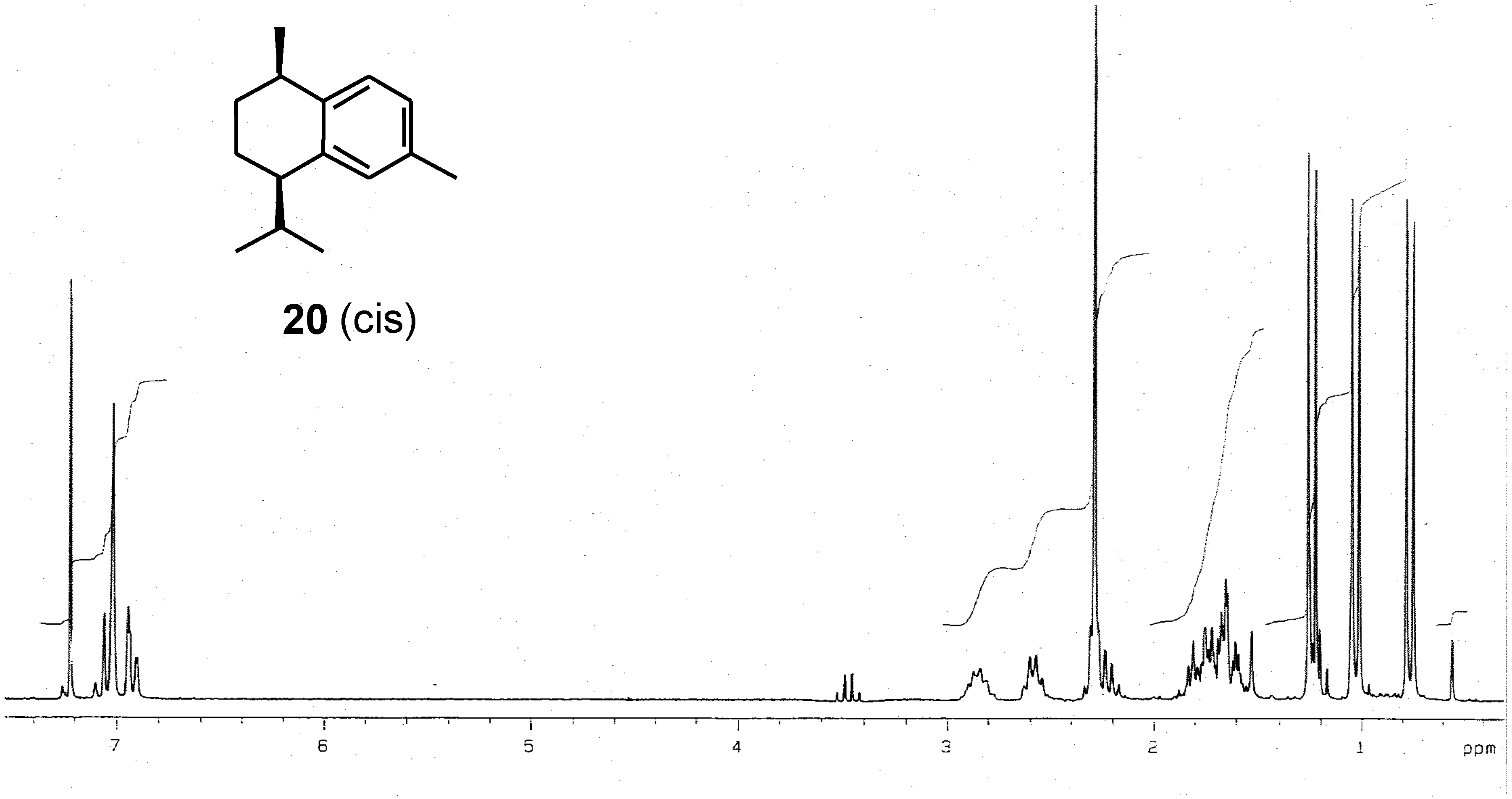
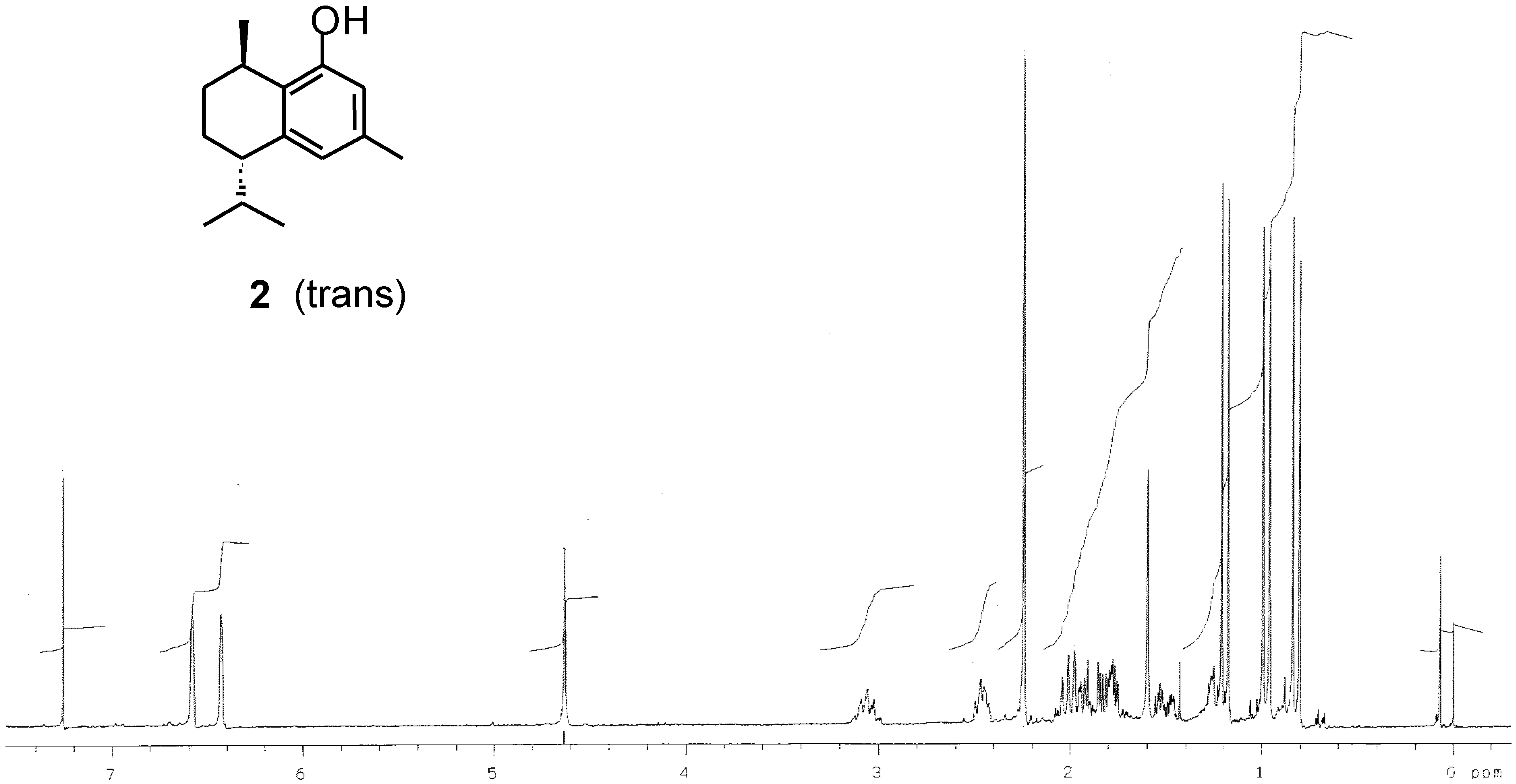
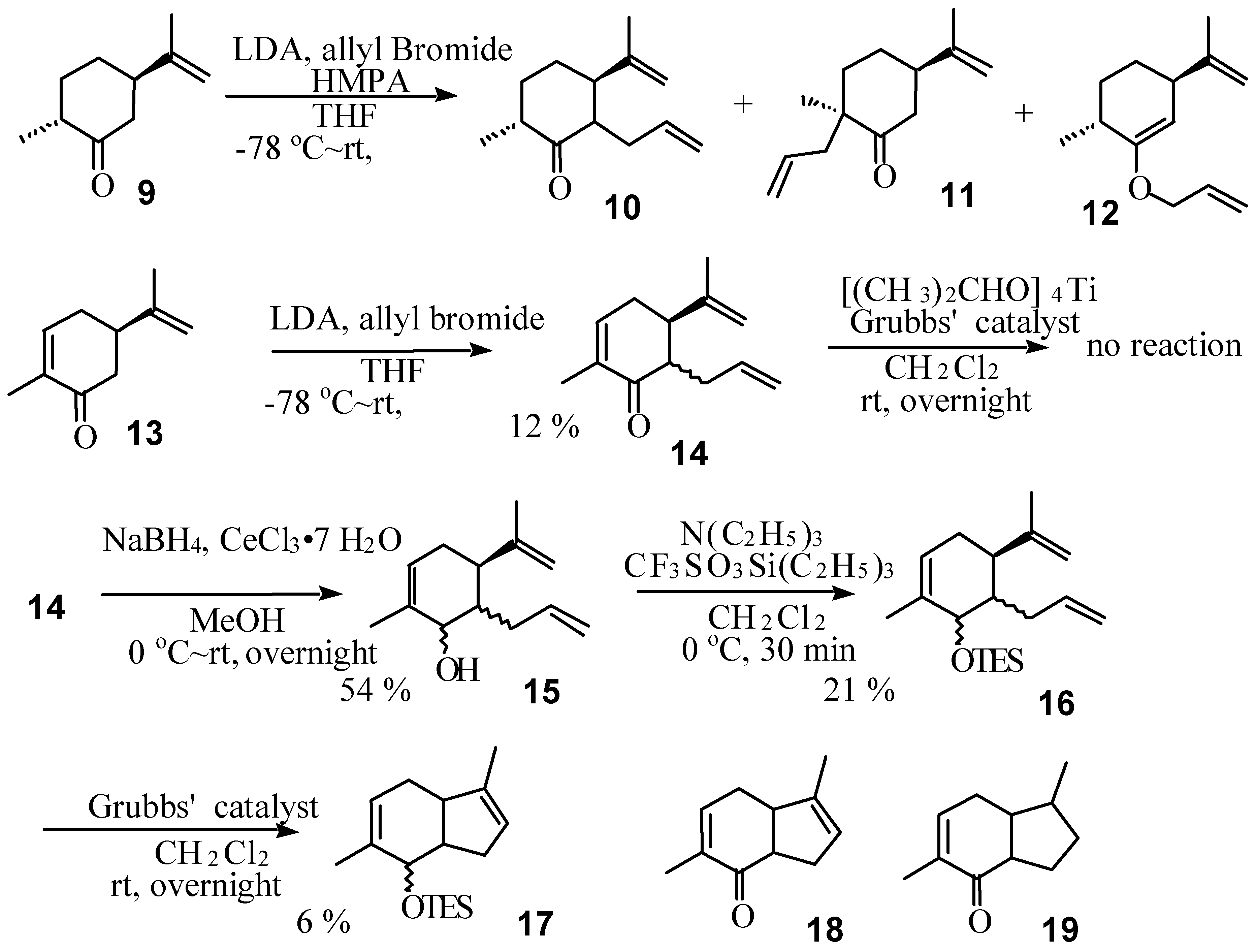
Results and Discussion
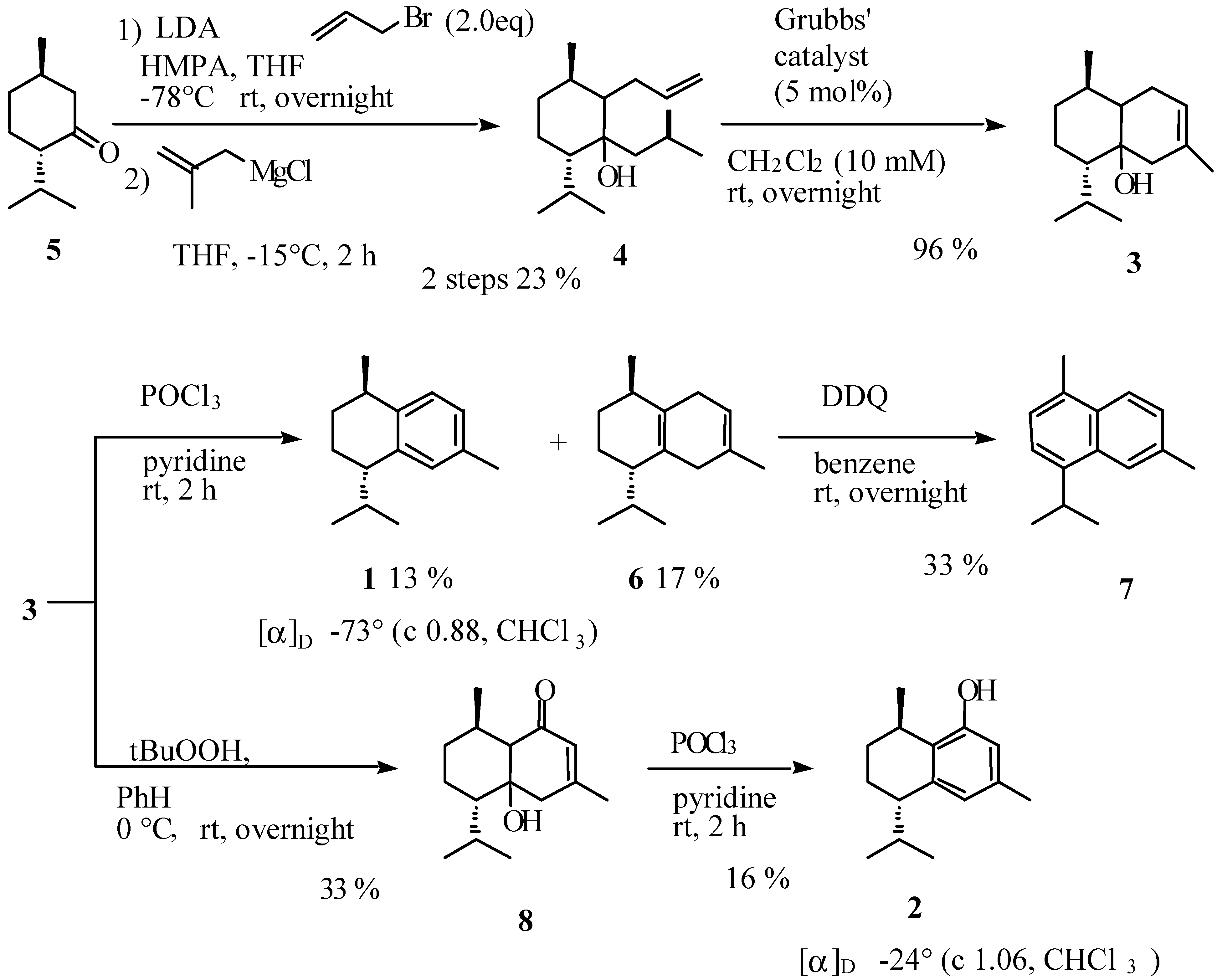

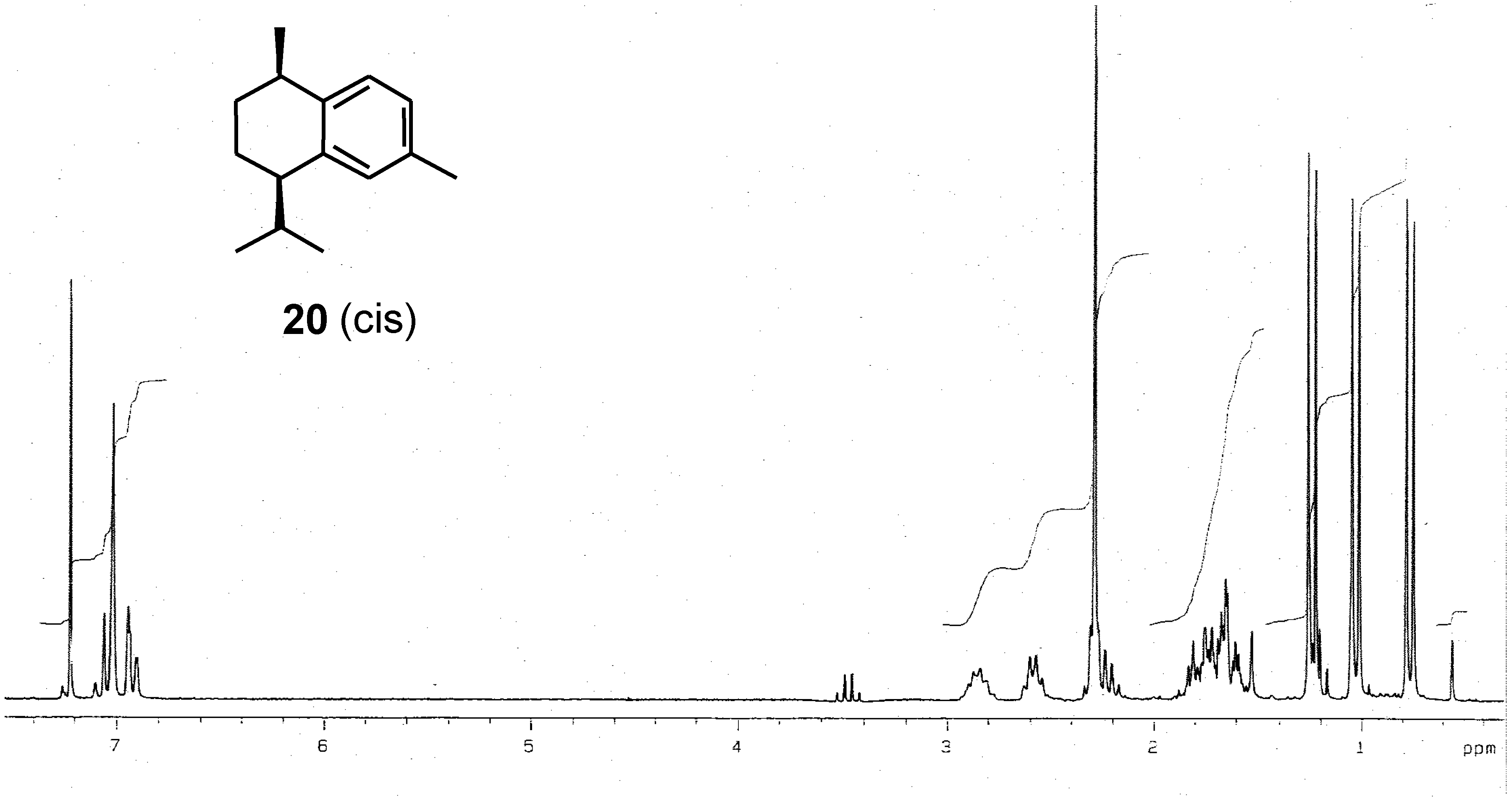

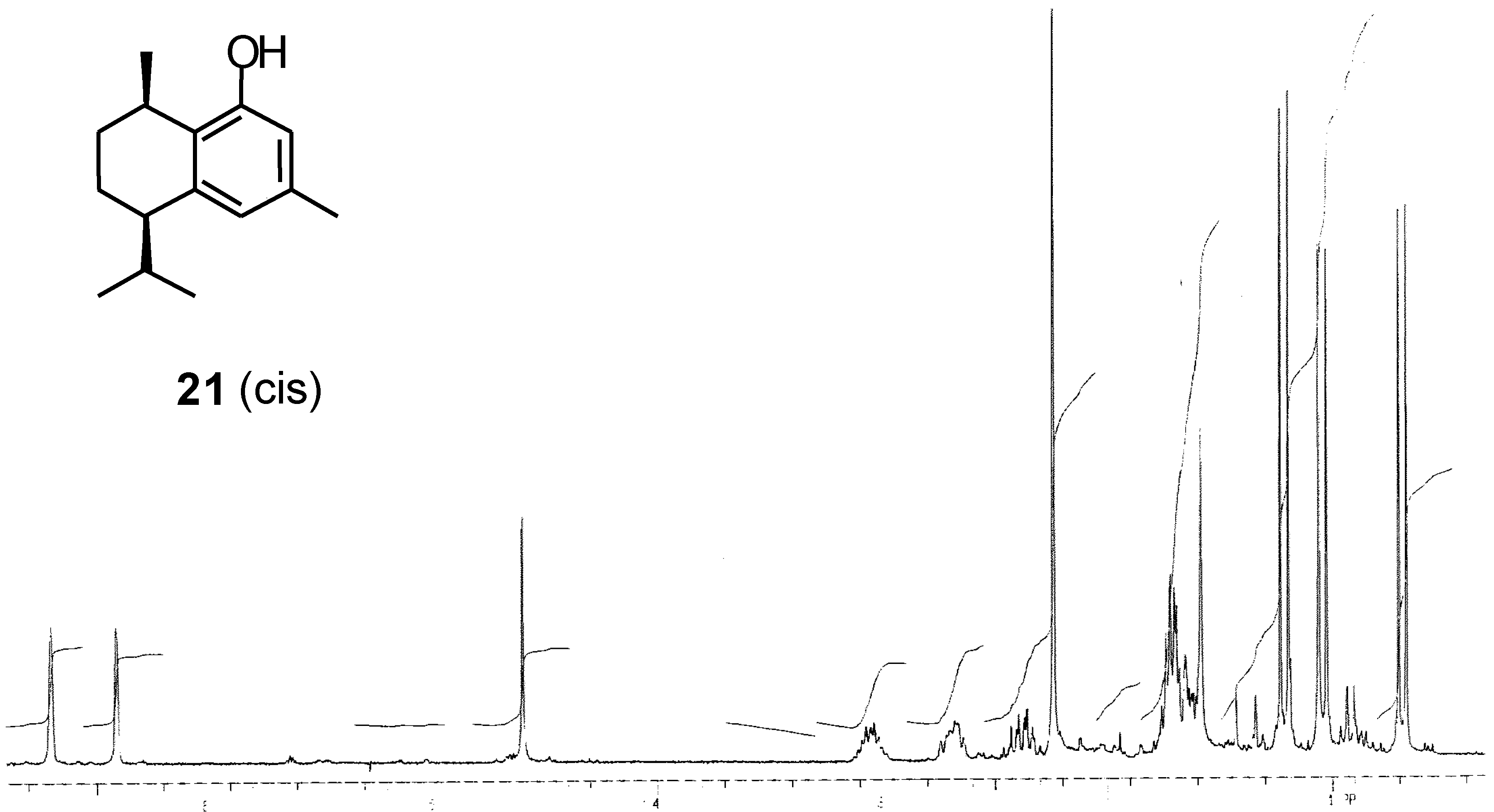
Conclusions

{kind=link}
{kind=link}
{kind=link}
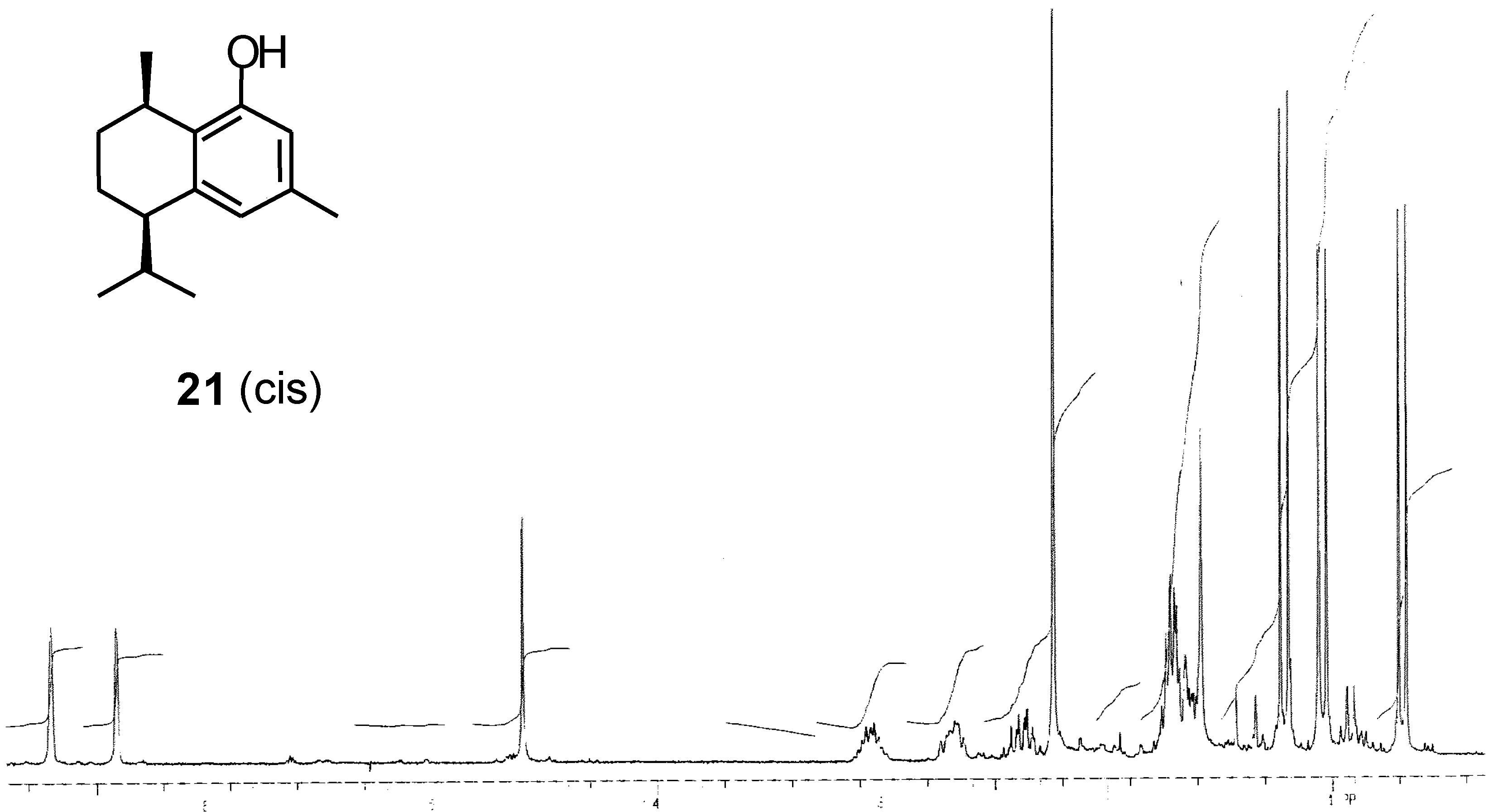{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound |  |  |  |  |
| 1 | ent-1 | 20 | ent-20 | |
| Synthetic product (this work) | -73 | |||
| Reported data | -96 [1] -77 [18] | +31 [7] | -22 [1, wrong] +43 [4] +33.4 [8] | -31 [17] |
| Compound |  |  |  |  |
| 2 | ent-2 | 21 | ent-21 | |
| Synthetic product (this work) | -24 | - | - | - |
| Reported data | - | +38 [3] | +40[20] | - |
| No. | 1 | 1 | 20 | 20 | 2 | ent-2 | 21 |
|---|---|---|---|---|---|---|---|
| this work* | [18] | [20]* | [18] | this work* | [3] | [20]* | |
| 1 | 140.1 | 140.3 | 139.7 | 140.0 | 126.1 | 126.5 | 126.7 |
| 2 | 126.8 | 127.0 | 128.5 | 128.5 | 153.2 | 153.2 | 153.0 |
| 3 | 126.2 | 126.4 | 126.3 | 126.5 | 113.4 | 113.8 | 112.8 |
| 4 | 134.5 | 134.6 | 134.5 | 134.6 | 135.1 | 135.1 | 135.7 |
| 5 | 128.8 | 129.0 | 128.7 | 128.9 | 123.0 | 123.1 | 120.8 |
| 6 | 140.0 | 140.3 | 139.9 | 140.2 | 141.3 | 141.3 | 141.3 |
| 7 | 43.8 | 44.1 | 43.6 | 43.8 | 43.0 | 43.2 | 43.3 |
| 8 | 21.1 | 21.3 | 23.3 | 23.3 | 19.1 | 19.2 | 16.4 |
| 9 | 30.8 | 31.0 | 28.7 | 28.9 | 27.1 | 27.2 | 28.8 |
| 10 | 32.5 | 32.6 | 31.1 | 31.2 | 26.5 | 26.7 | 26.5 |
| 11 | 31.9 | 32.1 | 32.5 | 32.7 | 33.1 | 33.2 | 30.9 |
| 12 | 17.3 | 17.5 | 17.6 | 17.8 | 19.6 | 19.7 | 17.3 |
| 13 | 21.5 | 21.6 | 21.4 | 21.5 | 21.0 | 21.1 | 20.5 |
| 14 | 22.3 | 22.3 | 19.7 | 22.5 | 22.1 | 22.2 | 21.2 |
| 15 | 21.3 | 21.3 | 21.2 | 21.2 | 21.1 | 21.3 | 21.1 |
Experimental
General
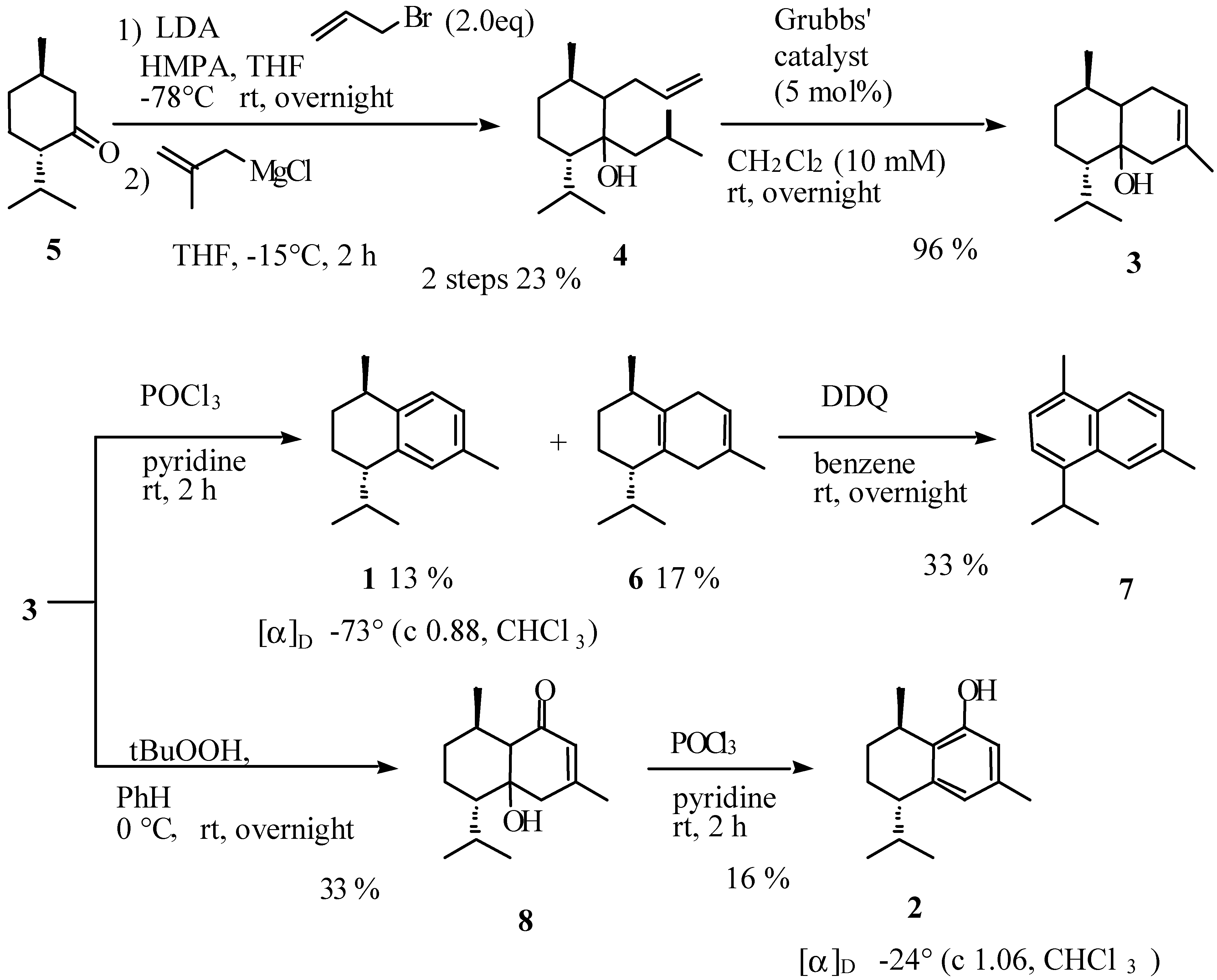
Preparation of [1ξ,2ξ,3R,6S]-2-allyl-6-isopropyl-3-methyl-1-(2-methylpropyl)cyclohexan-1-ol (4).
Preparation of [1ξ,6ξ,7R,10S]-10-isopropyl-3,7-dimethylbicyclo[4.4.0]dec-3-en-1-ol (3).
Preparation of (-)-calamenene (1) and [7S,10R]-2,5-dihydrocalamenene (6).
Preparation of [1ξ,6ξ,7S,10R]-6-hydroxy-7-isopropyl-4,10-dimethylbicyclo[4.4.0]dec-3-en-2-one (8).
Preparation of (-)-[7S,10R]-2-hydroxycalamenene (2).
Acknowledgements
References and Notes
- Andersen, N. H.; Syrdal, D. D.; Graham, C. The absolute stereochemistry of calamenene. Tetrahedron Lett. 1972, 905–908. [Google Scholar] [CrossRef]
- Connolly, J. D.; Hill, R. A. Dictionary of Terpenoids; Chapman & Hall: London, 1991. [Google Scholar] The name and numbering used followed those proposed in this source.
- Nishizawa, M.; Inoue, A.; Sastrapradja, S.; Hayashi, Y. (+)-8-Hydroxycalamenene: A fish-poison principle of Dysoxylum acutangulum and D. alliaceum. Phytochemistry 1983, 22, 2083–2085. [Google Scholar] [CrossRef]
- Croft, K. D.; Ghisalberti, E. L.; Hocart, C. H.; Jefferies, P. R.; Raston, C. L.; White, A. H. Absolute configuration of the (+)-calamenenes: Crystal structure of 7-hydroxycalamenene. J. Chem. Soc. Perkin 1 1978, 1267–1270. [Google Scholar] [CrossRef]
- Asakawa, Y. Progress in the Chemistry of Organic Natural Products; Herz, W., Grisebach, H., Kirby, G. W., Eds.; Springer-Verlag: Wien, 1982; Vol. 42, p. 1. [Google Scholar]
- Asakawa, Y. Progress in the Chemistry of Organic Natural Products; Herz, W., Kirby, G. W., Moor, R. E., Steglich, W., Tamm, Ch., Eds.; Springer-Verlag: Wien, 1995; Vol. 65, p. 1. [Google Scholar]
- Nagashima, F.; Suda, K.; Asakawa, Y. Cadinane-type sesquiterpenoids from the liverwort Scapania undulata. Phytochemistry 1994, 37, 1323–1325. [Google Scholar] [CrossRef]
- Nagashima, F.; Momosaki, S.; Watanabe, Y.; Takaoka, S.; Huneck, S.; Asakawa, Y. Sesquiterpenoids from the liverworts Bazzania trilobata and Porella canariensis. Phytochemistry 1996, 42, 1361–1366. [Google Scholar] [CrossRef]
- Ladwa, P. H.; Joshi, G. D.; Kulkarni, S. N. Stereochemical disposition of isopropyl group in synthetic (+)-calamenen. Indian J. Chem. 1978, 16B, 853–855. [Google Scholar]
- Nakashima, K.; Ito, R.; Sono, M.; Tori, M. Olefin metathesis reactions of some aromatic dienes with ortho- and meta-disubstitution. Formation of 10-, 12-, 14-, and 17-membered cyclic compounds and isomerization of an allylic alcohol. Heterocycles 2000, 53, 301–314. [Google Scholar] [CrossRef]
- Nakashima, K.; Imoto, M.; Miki, T.; Miyake, T.; Fujisaki, N.; Fukunaga, S.; Mizutani, R.; Sono, M.; Tori, M. Ring closing metathesis reaction of dienes with acrylate moiety leading to 5- to 7-membered lactones and cyclization to 14-membered rings. Heterocycles 2002, 56, 85–89. [Google Scholar] [CrossRef]
- Grubbs, R. H.; Chang, S. Recent advances in olefin metathesis and its application in organic synthesis. Tetrahedron 1998, 54, 4413–4450. [Google Scholar] [CrossRef]
- Fürstner, A. Alkene Metathesis in Organic Synthesis; Springer: Heidelberg, 1998. [Google Scholar]
- Fürstner, A. Olefin metathesis and beyond. Angew. Chem. Int. Ed. 2000, 39, 3012–3043. [Google Scholar] [CrossRef]
- Tori, M.; Sono, M.; Asakawa, Y. Absolute configuration and synthesis of the liverwort sesquiterpene alcohol tamariscol. J. Chem. Soc. Perkin Trans. 1 1990, 2849–2850. [Google Scholar] [CrossRef]
- Tori, M.; Sono, M.; Nishigaki, Y.; Nakashima, K.; Asakawa, Y. Studies on the liverwort sesquiterpene alcohol tamariscol. Synthesis and absolute configuration. J. Chem. Soc. Perkin Trans. 1 1991, 435–445. [Google Scholar] [CrossRef]
- Kim, Y. –K.; Cool, L. G.; Zavarin, E. cis-Calamenene-related sesquiterpenoids from Cupressus bakeri foliage. Phytochemistry 1994, 36, 961–965. [Google Scholar]
- Bunko, J. D.; Ghisalberti, E. L.; Jefferies, P. R. (1R,4S)-Calamenene. Chemical correlation with the diterpene analogues of Eremophila. Aust. J. Chem. 1981, 34, 2237–2242. [Google Scholar] [CrossRef]
- Toyota, M.; Asakawa, Y.; Takemoto, T. Sesquiterpenes from Japanese liverworts. Phytochemistry 1981, 20, 2359–2366. [Google Scholar] [CrossRef]
- Nagashima, F.; et al. unpublished data.
- Sample Availability: Samples are not available.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Nakashima, K.; Imoto, M.; Sono, M.; Tori, M.; Nagashima, F.; Asakawa, Y. Total Synthesis of (-)-(7S,10R)-Calamenene and (-)-(7S,10R)-2-Hydroxycalamenene by Use of a Ring-Closing Metathesis Reaction. A Comparison of the cis- and trans-Isomers. Molecules 2002, 7, 517-527. https://doi.org/10.3390/70700517
Nakashima K, Imoto M, Sono M, Tori M, Nagashima F, Asakawa Y. Total Synthesis of (-)-(7S,10R)-Calamenene and (-)-(7S,10R)-2-Hydroxycalamenene by Use of a Ring-Closing Metathesis Reaction. A Comparison of the cis- and trans-Isomers. Molecules. 2002; 7(7):517-527. https://doi.org/10.3390/70700517
Chicago/Turabian StyleNakashima, Katsuyuki, Masashi Imoto, Masakazu Sono, Motoo Tori, Fumihiro Nagashima, and Yoshinori Asakawa. 2002. "Total Synthesis of (-)-(7S,10R)-Calamenene and (-)-(7S,10R)-2-Hydroxycalamenene by Use of a Ring-Closing Metathesis Reaction. A Comparison of the cis- and trans-Isomers" Molecules 7, no. 7: 517-527. https://doi.org/10.3390/70700517