Synthesis and Theoretical Study of a New Type of Pentacyclic bis-Benzothiazolium Compound

Faculty of Natural Sciences, Comenius University, Mlynska dolina CH-2, SK-842 15, Bratislava, Slovakia
*
Author to whom correspondence should be addressed.
Molecules 2002, 7(7), 534-539; https://doi.org/10.3390/70700534
Submission received: 10 June 2002
/
Revised: 29 July 2002
/
Accepted: 30 July 2002
/
Published: 31 July 2002
Abstract
:The synthesis of a new type of pentacyclic benzothiazolium compound - 6,13-dihydropyrazino[2,1-b:5,4-b′]bis(1,3-benzothiazole)-7,14-diiumdibromide (2), is reported. Compound 2 was prepared by dimerization of 2-(bromomethyl)benzothiazole. Quantum chemical calculation studies have been carried out on the structures of possible isomers of 2, as well as the products of its deprotonation reactions.
Introduction
Many 2-substituted benzothiazole derivatives are reported to possess a broad spectrum of biological activities [1]. When quaternized to benzothiazolium salts, these compounds are especially active as antimicrobial [2], antihelmintic [3] and antineoplastic agents [4]. We are particularly interested in the study of the antimicrobial 3-alkylbenzothiazolium salts substituted at the 2-position by a conjugated fragment [5,6,7]. In this work we present the synthesis and theoretical study of a new type of pentacyclic benzothiazolium salt - 6,13-dihydropyrazino[2,1-b:5,4-b′]bis(1,3-benzothiazole)-7,14-diiumdibromide (2).
Results and Discussion
Chemistry
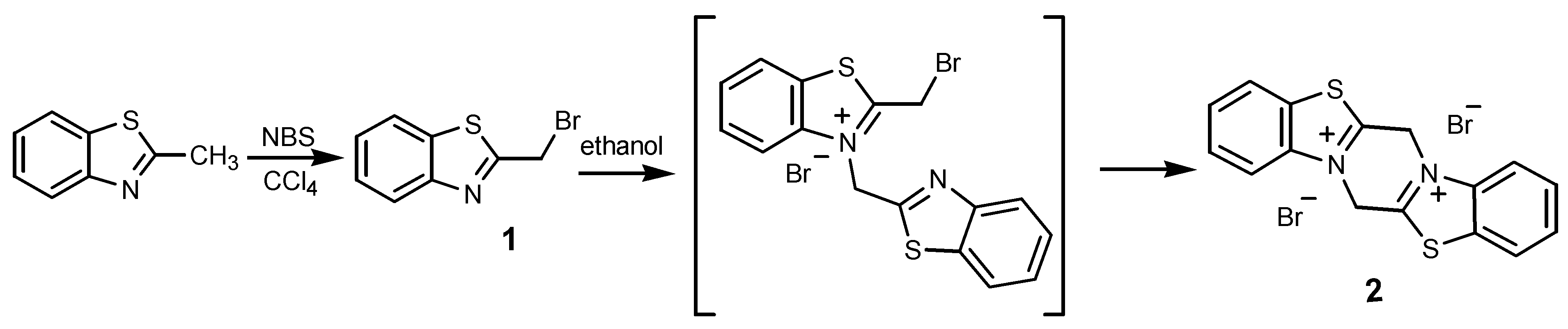
The starting material 2-(bromomethyl)benzothiazole (1) was obtained from commercially available 2-methylbenzothiazole by standard radical bromination in a nonpolar solvent [8]. The target product 2 was prepared by the autoalkylation reaction of compound 1 in dry ethanol (Scheme 1); reaction conditions such as temperature, reaction time and molar ratios of 2-(bromomethyl)benzothiazole to ethanol were optimized and the best conditions found are presented in the Experimental Section. This reaction probably proceeds via the monoalkylated compound 3-(benzothiazol-2-ylmethyl)-2-(bromomethyl)benzothiazolium bromide, which would explain the formation of the insoluble compounds in concentrated polar solutions upon mild warming due to the possibility of an intermolecular reaction of the monoalkylated derivative with another molecule of 2-(bromomethyl)benzothiazole. The solvolysis products 2-(ethoxymethyl)benzothiazole (yield 17 mol%), 2-(ethoxymethyl)-3H-benzothiazoliumbromide (yield 38 mol%) and 2-(bromomethyl)-3H-benzothiazolium bromide (yield 14 mol%) were also identified and isolated from the reaction mixture.
Scheme 1.

Theoretical study
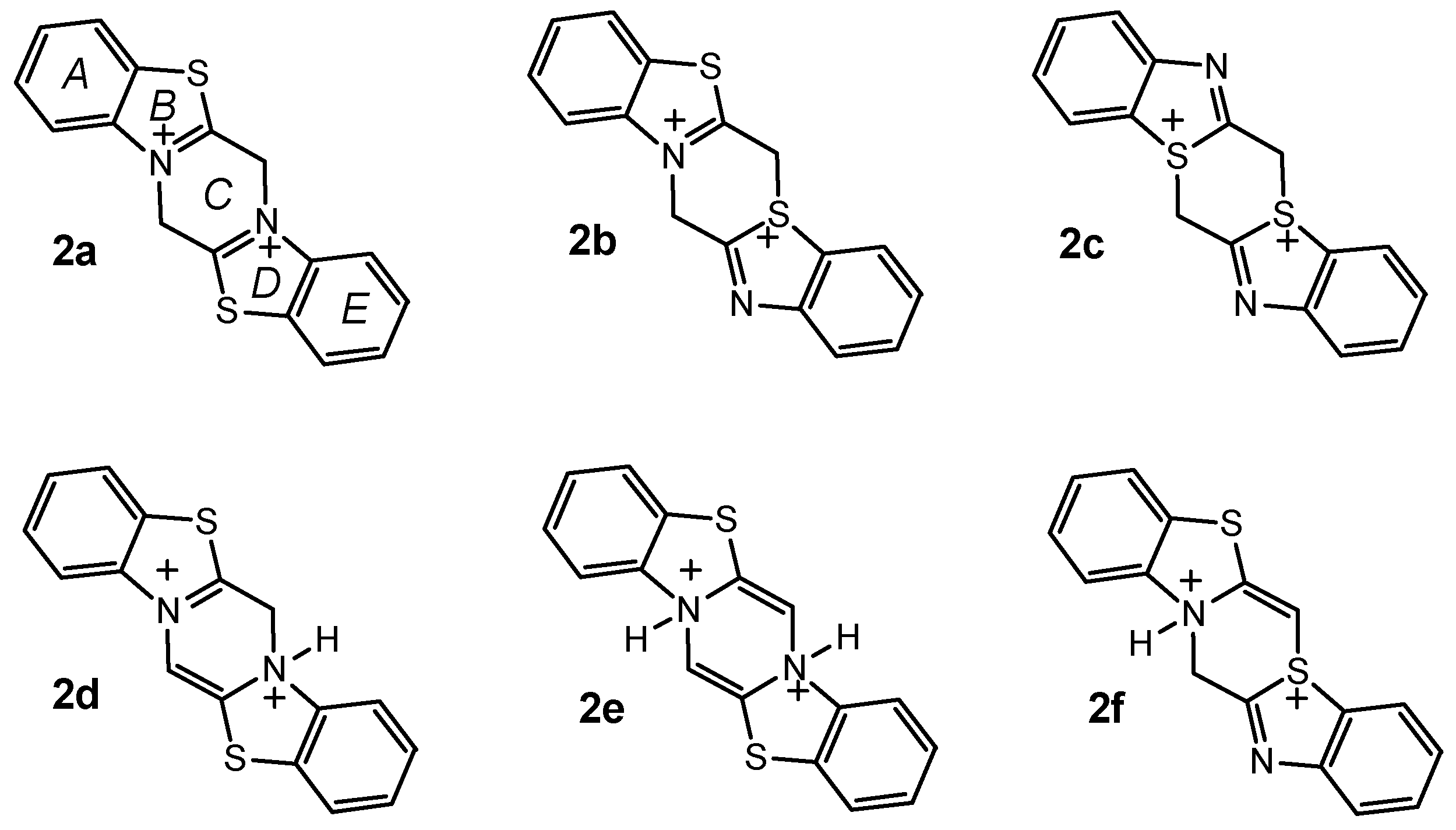
To obtain a better understanding of the mechanism of the reaction and the structure of the product, we carried out quantum chemical calculations using the semiempirical AM1 method [9]. The structures of the six isomeric dications 2a-f are presented in Scheme 2. The geometry of all isomers has been fully optimized. The ring nomenclature is shown for isomer 2a.
Scheme 2.

The calculated quantum-chemical data of isomers 2a-f is summarised in Table 1, where HF represents the heat of formation, ΔE is the relative stability calculated as a difference between heats of formation of the corresponding isomer and the most stable isomer 2a, EHOMO is the energy of the highest occupied molecular orbital and ELUMO is the energy of the lowest unoccupied molecular orbital.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HF [kcal/mol] | ΔE [kcal/mol] | EHOMO [eV] | ELUMO [eV] |
|---|---|---|---|---|
| 2a | 490.6 | 0.0 | -15.83478 | -8.48669 |
| 2b | 512.0 | 21.4 | -15.86577 | -8.95737 |
| 2c | 528.1 | 37.5 | -16.31676 | -9.21274 |
| 2d | 515.9 | 25.3 | -14.83067 | -9.15739 |
| 2e | 556.2 | 65.6 | -15.38245 | -8.46445 |
| 2f | 536.0 | 45.4 | -15.54802 | -8.76327 |
As it can be seen from the data in Table 1, the most stabilizing structural element is the planar tetravalent nitrogen atom with one double and two single bonds common to two heterocycles which occurs in isomers 2a, 2b and 2d. The remaining structural elements, namely the tetravalent nitrogen with three single N-C bonds and one N-H bond as well as the trivalent sulphur with three S-C bonds, are not planar and destabilize the molecule.
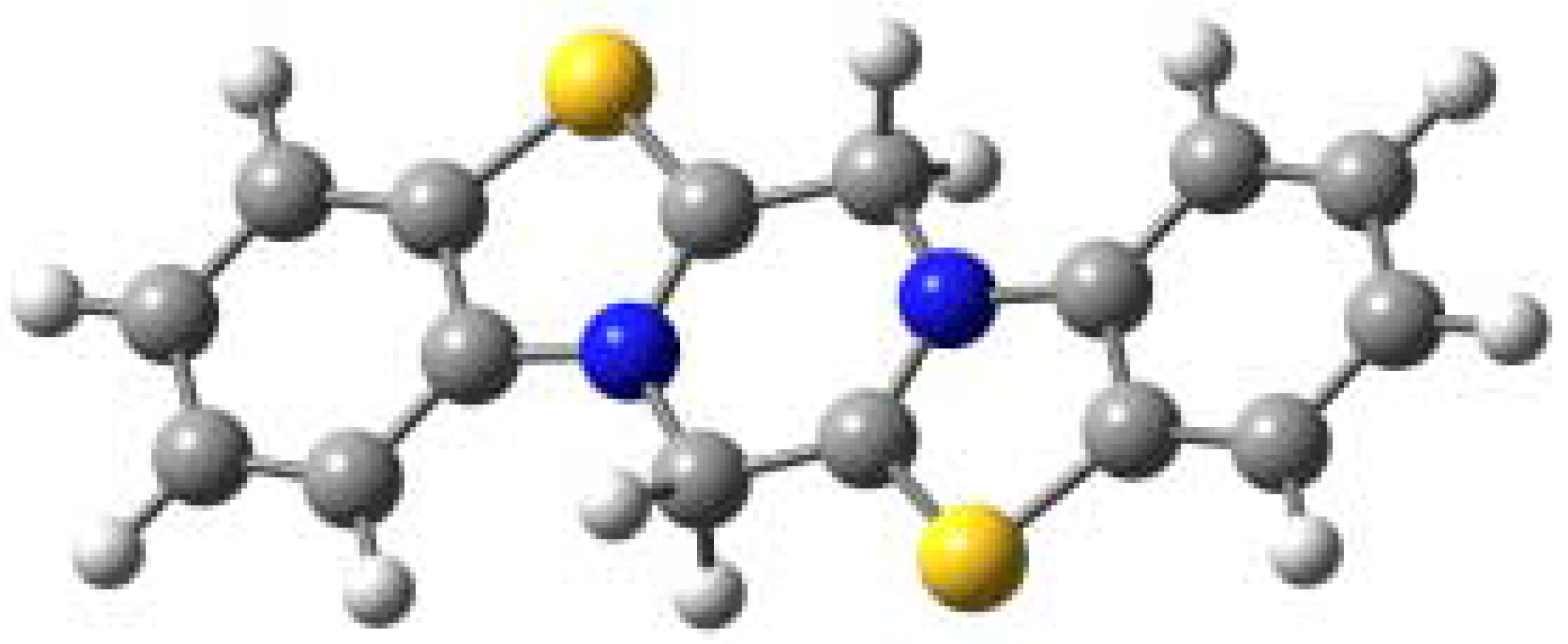
The AM1 calculated optimized geometry of the most stable isomer 2a is shown in Figure 1. This isomer is symmetric with two nitrogen atoms common to two rings and the molecule is nearly planar (the maximum out of plane deformation is about 4° in the ring C).
Figure 1.
Calculated structure of dication 2a (
, )


The symmetry of the molecule is also evident from the charge densities of the atoms. The sp3 hybridized carbons in ring C are slightly deformed from a tetrahedral arrangement with an H-C-H angle of 108.5° and a C-C-N angle of 113.6°. The preference for structure 2a is further confirmed by the 1H-NMR spectra, which are consistent with a symmetric structure with four equivalent hydrogens in ring C. As for the remaining data, the minimum frontier orbital energy corresponds to isomer 2c, the maximum EHOMO to 2d and ELUMO to 2e.
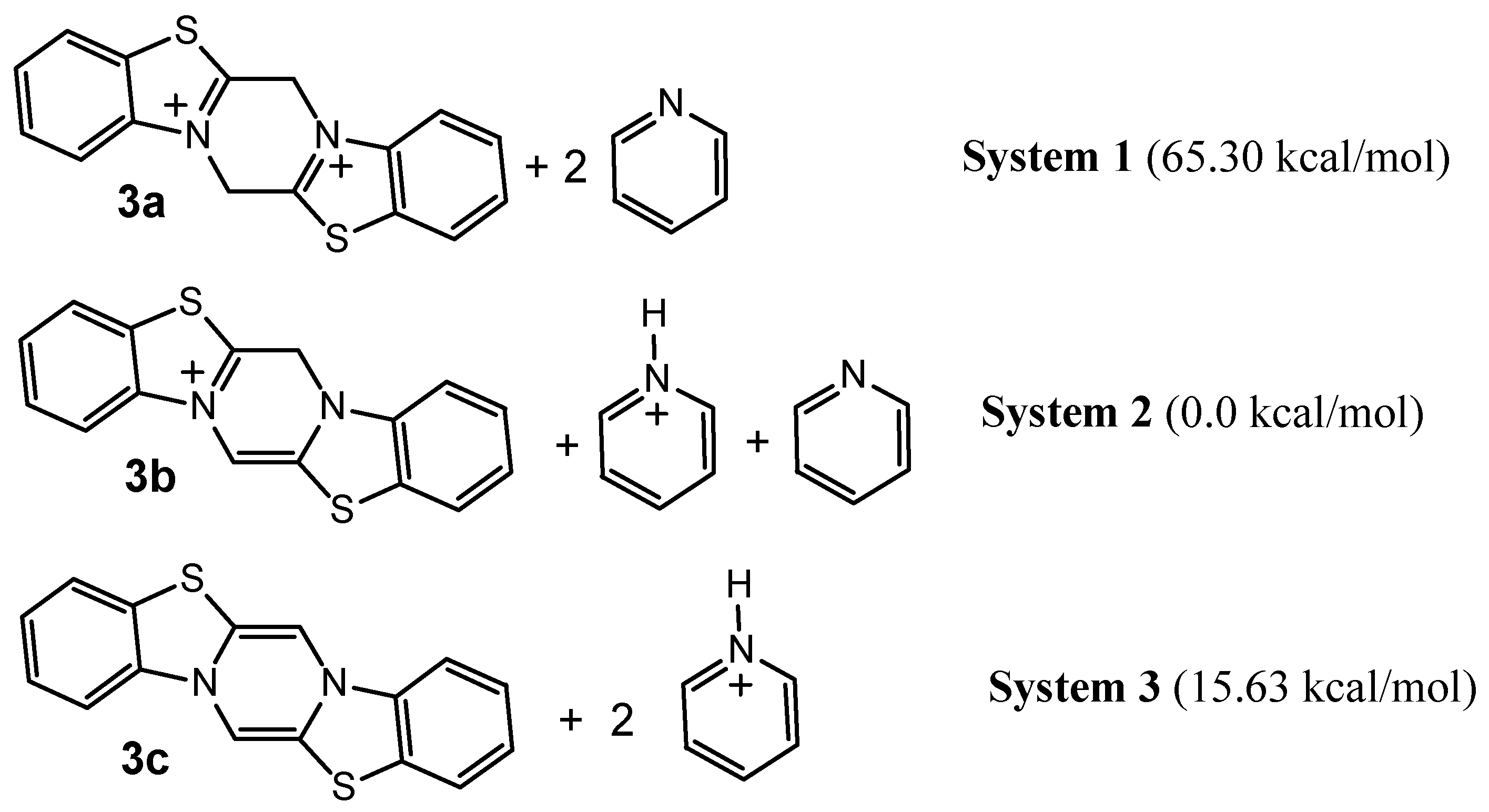
As we plan to study the condensation reactions of compound 2a, a theoretical study of the deprotonation of 2a was our next goal. For this purpose we modeled the reaction of 2a with pyridine as a base. To compare the energy of deprotonation, the following isodesmic reactions with pyridine as a model base were considered (Scheme 3). The value of the energy is the relative stability of the system defined as the difference Ex-E2, where Ex is the sum of heats of formation of three molecules in the corresponding system (System 3 or System 1) and E2 is the sum of heats of formation of molecules in the most stable System 2.
Scheme 3.

The energy data in Scheme 3 show that System 2 is the most stable one, followed by System 3 and then the dication 3a with two pyridine molecules (System 1). This means that the deprotonation of the C ring in 3a by the pyridine molecule ( System 1 → System 2) produces a 65.30 kcal/mol energy decrease. On the contrary, removal of the proton bonded to the sp3-carbon in ring C of monocation 3b ( System 2 → System 3) is an endothermic process with a 15.63 kcal/mol increase in energy. This fact is confirmed also by the calculated charge densities: the hydrogen in ring C of dication 3a is more acidic (Q = + 0.176) than the hydrogen bonded to the sp3-carbon in ring C of monocation 3b (Q = + 0.132). The optimisation of conditions for the condensation reactions of 3a is currently in progress.
Experimental
General
Melting points were determined on the Kofler block and are uncorrected. Elemental analyses were determined on a Carlo Erba 1016 instrument. 1H-NMR spectra were recorded with a 300MHz Varian Gemini 2000 instrument using TMS as an internal standard. Column chromatography was performed using silica gel (40/100 mesh), thin-layer chromatography was performed on silica Silufol UV-254 precoated plates (Kavalier, Czech Republic).
Synthesis of 2-(bromomethyl)benzothiazole (1).
The 2-(bromomethyl)benzothiazole starting material was synthesized by a standard procedure [8]. A stirred mixture of 2-methylbenzothiazole (0.1mol, 15g), N-bromosuccinimide (0.056mol, 9g) and azobis(isobutyronitrile) (1.83mmol, 0.3g) in dry CCl4 (150mL) was slowly warmed to 70°C and then stirred for 2h at this temperature. The resulting suspension was filtered off, washed with CCl4 and solvent was evaporated under reduced pressure. The reaction product was purified by flash chromatography (silica gel, 5:1 hexane - ethyl acetate) to give the expected product 1 (6.68g, 58%) as sticky white powder (WARNING: irritant!), m.p. 82°C (decomp.); Anal. Calculated for C8H6NSBr: 42.12% C, 2.65% H, 6.14% N, 14.05% S, 35.03% Br; Found: 42.21% C, 2.69% H, 6.07% N, 13.98% S, 34.90% Br; 1H-NMR (CDCl3): 8.01 (d, 1H, J4,5 = 7.4Hz, H-4); 7.88 (d, 1H, J6,7 = 8.1Hz, H-7); 7.50 (dd, 1H, J6,7 = 8.1Hz, J5,6 = 7.1Hz, H-6); 7.42 (dd, 1H, J4,5 = 7.4Hz, J5,6 = 7.1Hz, H-5); 4.82 (s, 2H, H-8). Atom numbering is shown in Scheme 4.
Scheme 4.

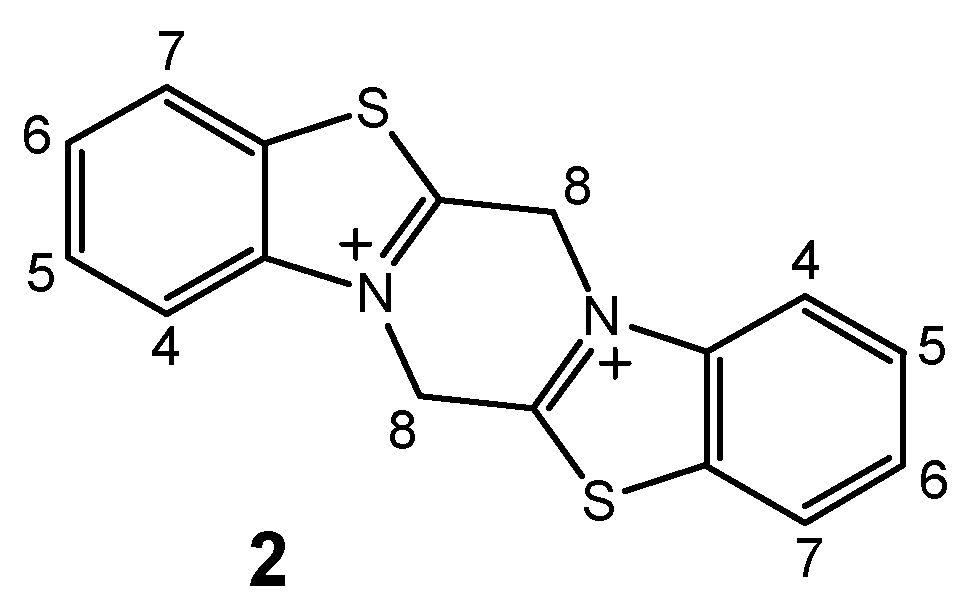
6,13-dihydropyrazino[2,1-b:5,4-b´]bis(1,3-benzothiazole)-7,14-diiumdibromide (2).
A solution of compound 1 (5.7mmol, 1.3g) in dry ethanol (300mL) was heated at 75°C for 4 days under nitrogen. The solvent was then evaporated under reduced pressure (without warming) and the residual orange oil was stirred in dry diethyl ether at room temperature for 20h to precipitate the solid product, which was then filtered off, washed with dry diethyl ether (5mL), resuspended in CHCl3 (30mL) and stirred at room temperature for 24h. The suspension was filtered again and the solids washed with CHCl3 (5mL) and resuspended in CH2Cl2 (30 mL) at room temperature for 24h. The suspension was again filtered and the solids washed with CH2Cl2 (5 mL) and dried in vacuo to give 0.13g (10%) of product 2, m.p.115°C (decomp.); Anal. Calculated for C16H12N2S2Br2: 42.12% C, 2.65% H, 6.14% N, 14.05% S, 35.03% Br; Found: 41.88% C, 2.72% H, 6.02% N, 13.87% S, 35.69% Br; 1H-NMR (2:1 CF3COOD-D2O): 8.16 (d, 2H, J6,7 = 8.1Hz, H-7); 8.13 (d, 2H, J4,5 = 8.4Hz, H-4); 7.89 (dd, 2H, J4,5 = 8.4Hz, J5,6 = 7.9Hz, H-5); 7.82 (dd, 2H, J6,7 = 8.1Hz, J5,6 = 7.9Hz, H-6); 5.48 (s, 4H, H-8). Atom numbering is presented in Scheme 5.
Scheme 5.

Quantum Chemical Calculation Methods
Acknowledgements
This work was supported by the Grant Agency of Science of Ministry of Education of the Slovak Republic (Grant No. 1/8207/01).
References
- Grandolini, G.; Ambrogi, V.; Rossi, C. Eur. J. Med. Chem. 1996, 21, 455.
- Afsah, S.; Nayyer, S. Asian J. Chem. 1988, 8, 419.
- Garmaise, D. L.; Paris, G. I.; Komlossy, J. J. Med. Chem. 1968, 12, 30.
- Krieg, M.; Bilitz, J. Biochem. Pharmacol. 1996, 51, 1461. [PubMed]
- Zahradník, P.; Foltínová, P.; Halgaš, J. SAR and QSAR in Envir. Res. 1996, 5, 51.
- Magdolen, P.; Zahradník, P.; Foltínová, P. Die Pharmazie 2000, 55, 803. [PubMed]
- Buffa, R.; Zahradnik, P.; Foltinova, P. Heterocycl. Commun. 2001, 7, 331.
- Sawyer, J.S.; Baldwin, R.F.; Rinkema, L.E.; Roman, C.R. J. Med. Chem. 1992, 35, 1200. [PubMed]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. J. Am. Chem. Soc. 1995, 107, 3902.
- AMPAC 6.0, 1997; Semichem, Shawnee Mission, KS 66222, U.S.A.
- Sample availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Zahradnik, P.; Buffa, R. Synthesis and Theoretical Study of a New Type of Pentacyclic bis-Benzothiazolium Compound. Molecules 2002, 7, 534-539. https://doi.org/10.3390/70700534
AMA Style
Zahradnik P, Buffa R. Synthesis and Theoretical Study of a New Type of Pentacyclic bis-Benzothiazolium Compound. Molecules. 2002; 7(7):534-539. https://doi.org/10.3390/70700534
Chicago/Turabian StyleZahradnik, Pavol, and Radovan Buffa. 2002. "Synthesis and Theoretical Study of a New Type of Pentacyclic bis-Benzothiazolium Compound" Molecules 7, no. 7: 534-539. https://doi.org/10.3390/70700534