Novel Heteroatom-containing Vitamin D3 Analogs: Efficient Synthesis of 1α,25-Dihydroxyvitamin D3-26,23-lactam

Institute of Molecular and Cellular Biosciences, University of Tokyo, Yayoi, Bunkyo-ku, Tokyo 113-0032, Japan
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(6), 488-499; https://doi.org/10.3390/80600488
Submission received: 13 May 2003
/
Revised: 20 May 2003
/
Accepted: 21 May 2003
/
Published: 30 June 2003
(This article belongs to the Special Issue Selected Papers from the Second Eurasian Meeting on Heterocyclic Chemistry: Heterocycles in Organic and Combinatorial Chemistry)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Vitamin D3 and its synthetic analogs are promising compounds for controlling various types of cell differentiation. In this article, we describe the synthesis of novel vitamin D3 analogs containing heteroatoms in their side chains – so-called vitamin D3 lactam analogs – via 1,3-dipolar cycloaddition reaction as a key step.
Introduction
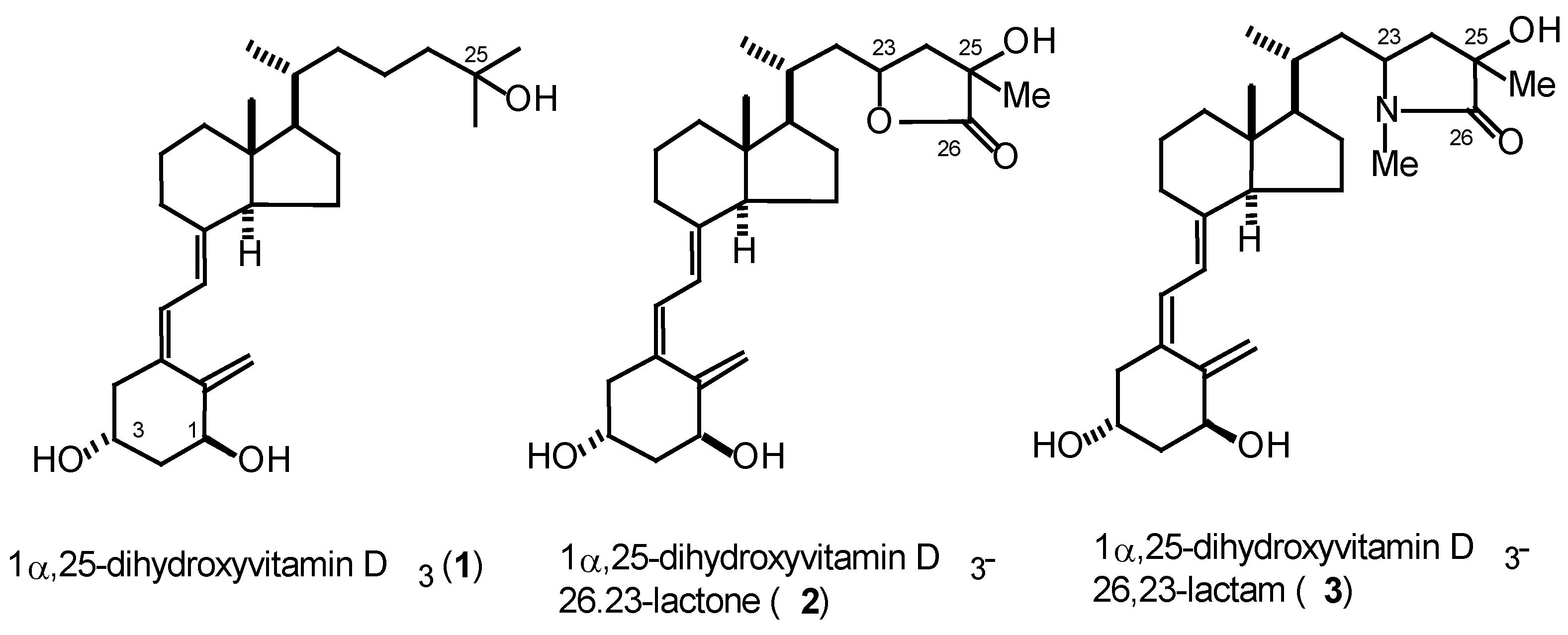
1α,25-Dihydroxyvitamin D3 (1) has a wide variety of biological activities including the regulation of calcium homeostasis and the control of cellular growth, differentiation and apoptosis [1]. Though many analogs of 1 have been synthesized so far, only a few analogs containing nitrogen atoms in their structures have been reported. In the course of our recent studies towards the development of novel vitamin D3 analogs, we focused on one of the major metabolites of 1, so-called (23S, 25R)-1α,25-dihydroxyvitamin D3-26,23-lactone (2a) which inhibits bone resorption induced by 1, while it has very weak binding affinity against vitamin D nuclear receptor [2]. Interestingly, each of the diastereomers of 2a, that are (23S,25S)-(2b), (23R,25R)-(2c), and (23R,25S)-(2d), has different biological activities. On the basis of the structure of 2, we have designed 1α,25-dihydoroxyvitamin D3-26,23-lactam (3) as a novel vitamin D3 analog. Herein we report the efficient synthesis of (23S,25R)-1α,25-dihydroxyvitamin D3-26,23-lactam (3a) including its A-ring and CD-ring synthons.
Figure 1.
Results and Discussion
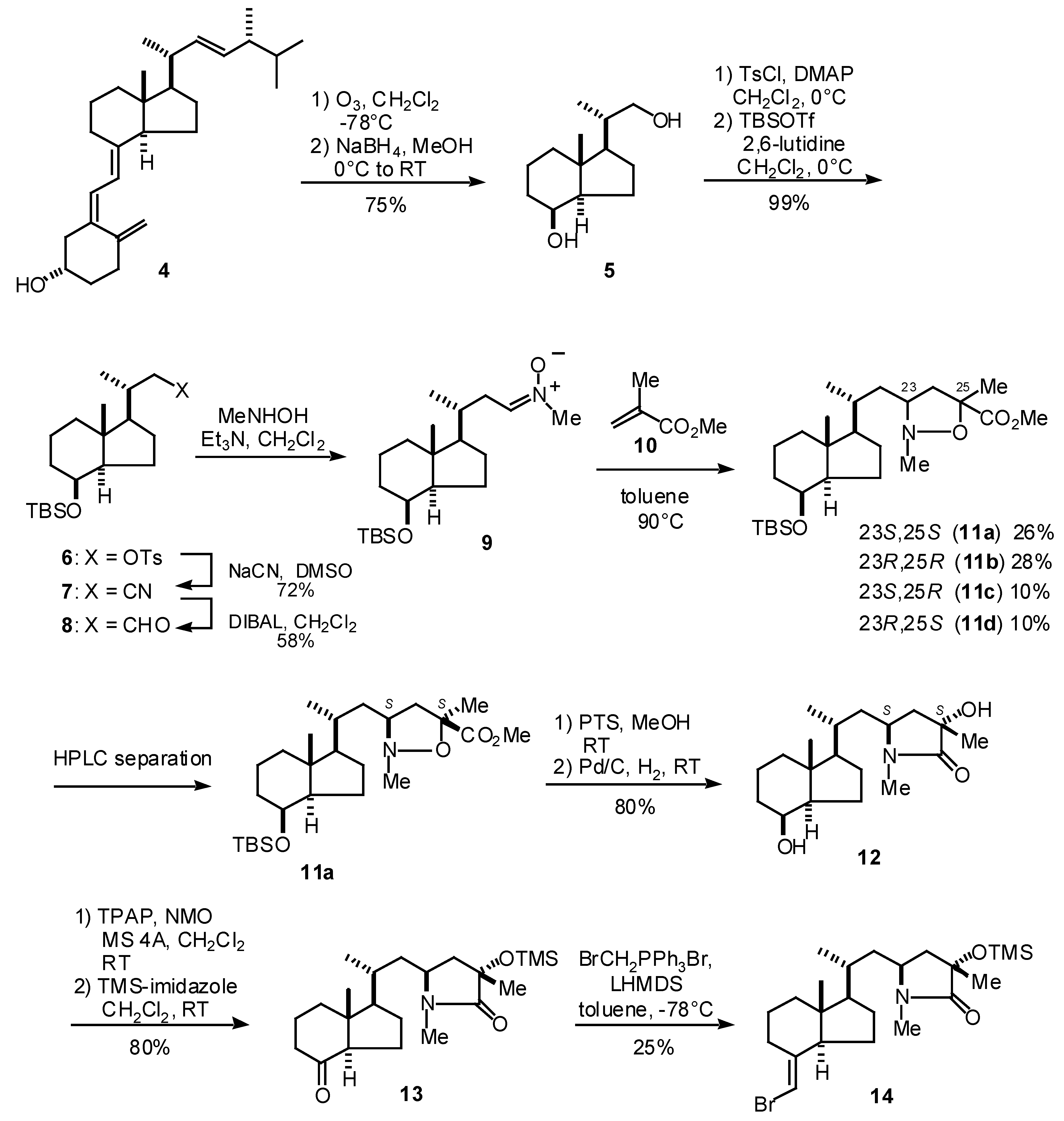
1. Synthesis of CD-ring synthon 14
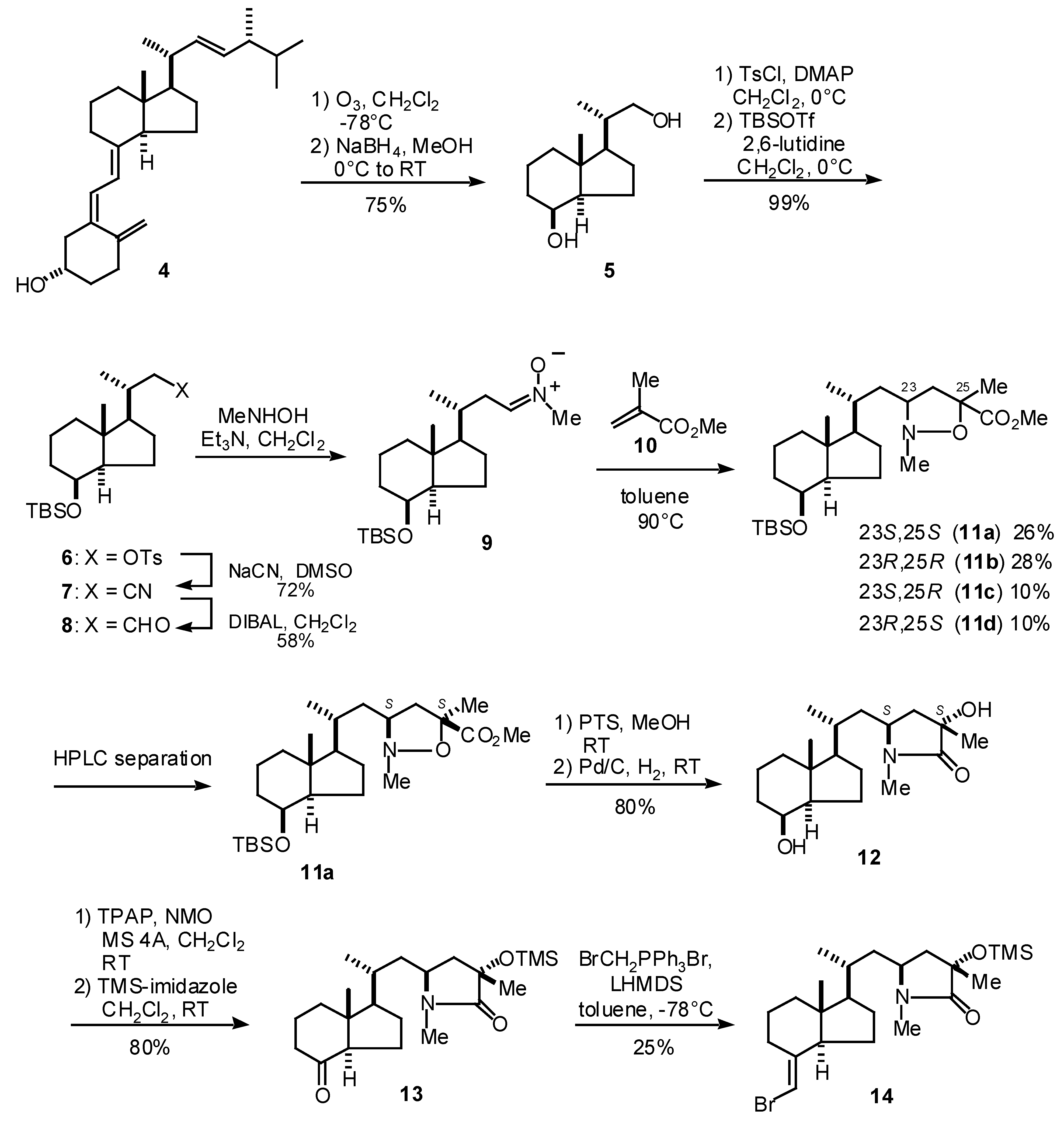
The synthesis of CD-ring synthon 14 for 3a is summarized in Scheme 1. The reaction of the Inhoffen-Lythgoe diol (5), prepared from vitamin D2 (4), with p-toluenesulfonyl chloride and DMAP followed by protection of the secondary alcohol with TBSOTf and 2,6-lutidine gave silyl ether 6 in 75% yield (2 steps). The tosylate 6 was converted into the aldehyde 8 with the following stepwise reaction: 1) cyanide formation with sodium cyanide and 2) reduction of the resulting cyanide with DIBAH [3]. The aldehyde 8 was reacted with N-methylhydroxy amine to give nitrone 9, which was subsequently reacted with methyl methacrylate (10) to give the four possible diastereomers of isoxazolidine 11. These diastereomers were separated with HPLC and 11a, 11b, 11c and 11d were thus obtained in 26, 28, 10 and 10% yields, respectively [4]. The stereochemistries of these compounds were confirmed by NMR techniques. One of the isomers, 11a, which has (23S,25S)-stereochemistry, was then carried on to the CD-ring synthon 14. Thus, the removal of the TBS group of 11a with p-toluenesulfonic acid in methanol and reduction of the N-O bond with hydrogen in the presence of palladium on carbon gave lactam 12 in 80% yield. The secondary alcohol of 12 was oxidized with TPAP-NMO [5] conditions and tertiary alcohol was protected with TMS ether by TMS-imidazole to give ketone 13 in 80%. Finally, the Wittig reaction of 13 under the Trost’s conditions [6] gave vinyl bromide 14 in 25% yield while the ketone 13 was recovered in 52%.
Scheme 1.
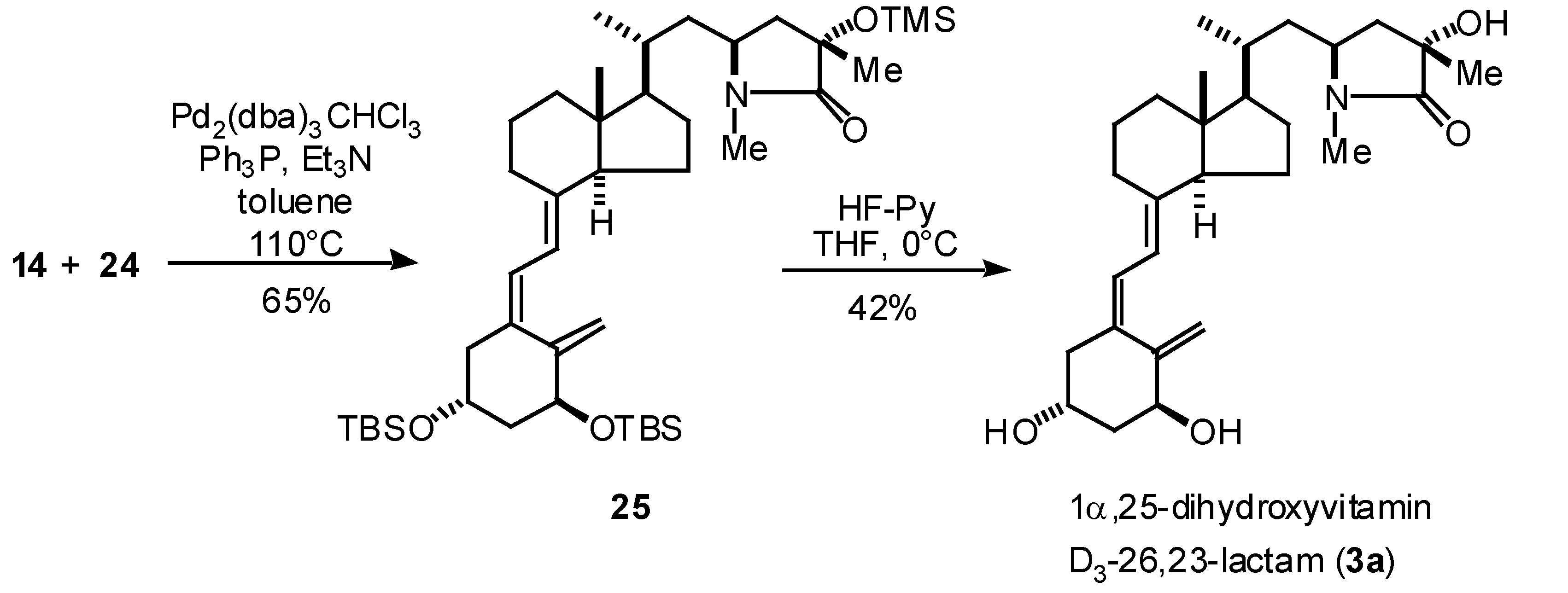
2. Synthesis of A-ring synthon 24 and total synthesis of (23S,25S)-1α,25-dihydroxyvitamin D3-26,23-lactam (3a)
The A-ring synthon 24 was synthesized from (S)-malic acid (15) (Scheme 2). The reduction of 15 with borane-methyl sulfide complex and following protection of the diol with p-anisaldehyde dimethylacetal and camphorsulfonic acid gave alcohol 17 in 59% yield. After oxidation of the primary alcohol of 17 under Swern conditions, the aldehyde generated was reacted with the methylene Wittig reagent, prepared from methyltriphenylphosphonium bromide and n-butyl lithium, to give 18 in 73% yield. The p-methoxybenzylidene acetal of 18 was selectively reduced with DIBAH at 0°C [7] gave alcohol 19 in 70% yield. The oxidation of the primary alcohol of 19 with Dess-Martin reagent [8] followed by propargylation with propargyl bromide and magnesium in the presence of a catalytic amount of mercury chloride gave 21 in 67% yield as a 1:1 mixture of diastereomers. The reaction of 21 with DDQ and subsequent purification on silica gel column gave deprotected diol 23a via acetal 22a and acetal 22b in 25% and 26% yield, respectively. The diol 23a was reacted with TBSOTf and 2,6-lutidine gave silyl ether 24 in 99% yield.
Scheme 2.

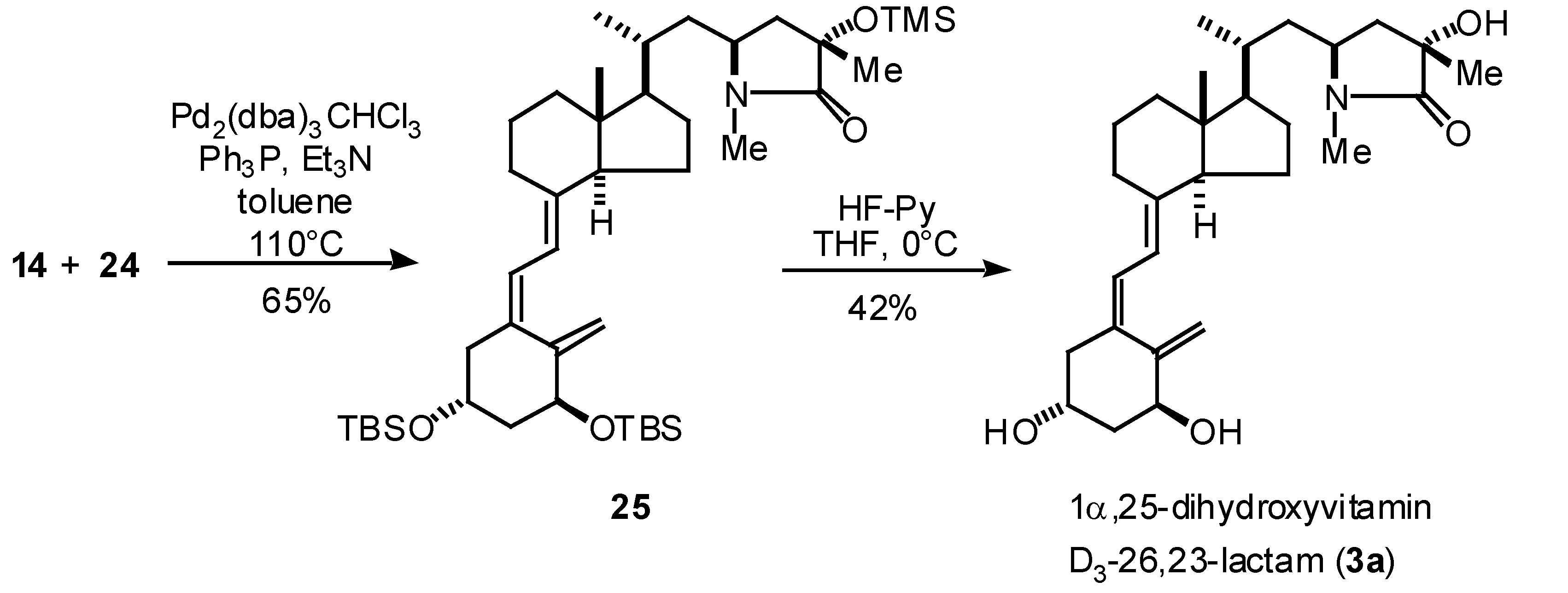
With the vinyl bromide 14 (CD-ring synthon) and eneyne 24 (A-ring synthon) in hand, coupling reaction of these segments were examined based upon the palladium catalyzed reaction conditions [6,9]. Coupling reaction of 14 and 24 in the presence of catalytic amount of Pd2(dba)3CHCl3 and triphenylposphine in toluene-triethylamine under the reflux conditions gave 25 in 65% yield. Deprotection of the hydroxyl protecting group was finally performed with HF-pyridine to give (23S,25S)-1α,25-dihydroxyvitamin D3-26,23-lactam (3a) in 42% yield.
Scheme 3.
Conclusions
In summary, we have designed and synthesized the novel vitamin D3 analog 3a having the lactam moiety on its CD-ring side chain. Our synthesis of 3a features: 1) 1,3-dipolar cycloaddition reaction between 9 and 10, followed by the reduction of an isoxazolidine to form the lactam moiety for 14 and 2) palladium catalyzed coupling reaction of CD-ring synthon 14 and A-ring synthon 24. Since this method provides a variety of vitamin D3 lactam analogs, synthetic efforts towards these, including (23R,25R)-(3b), (23R,25S)-(3c), and (23S,25R)-(3d), continue. Furthermore, the evaluation of the biological activities of synthetic analogs 3 in our laboratories is planned.
Experimental
General
Optical rotations were recorded with a JASCO DIP-370 polarimeter. IR spectra were measured with a JASCO VALOR-III FT-IR spectrophotometer. 1H- and 13C-NMR spectra were recorded on JNM-α500 instruments at 500 and 125 MHz, respectively. Mass spectra were recorded on JEOL JMA-HX110 spectrometers.
(8S,20R)-De-A,B-8-Hydroxy-20-(hydroxymethyl)pregnane (5). Vitamin D2 (4) (5.00 g, 12.63 mmol) was dissolved in CH2Cl2 (170 mL), and MeOH (70 mL) was added. After cooling to –78°C, a stream of ozone was passed to the solution for 1 h. The remaining ozone was purged with a stream of N2. After warming up to 0°C, NaBH4 (1.43g, 37.8 mmol) was added and stirred for 1h. The reaction mixture was then acidified by adding 2N HCl and extracted with CH2Cl2. The organic layer was washed with water, dried over MgSO4, filtered and concentrated in vacuo. The residue was chromatographed on silica gel (6:1 hexane - ethyl acetate) to give diol 5 (2.8g, 75%) as a white solid. 1H-NMR (CDCl3) δ 4.09 (d, J = 2.6 Hz, 1H), 3.64 (dd, J = 3.3, 10.6 Hz, 1H), 3.39 (dd, J = 6.6, 10.6 Hz, 1H), 1.99 (dd, J = 2.6, 13.2 Hz, 1H), 1.12~1.92 (m, 14H), 1.03 (d, J = 6.6 Hz, 3H), 0.002 (s, 3H).
(8S,20R)-De-A,B-8-(tert-Butyldimethylsilyl)oxy-20-[(p-tolylsulfonyl)oxymethyl]pregnane (6). DMAP (3.7 g, 30.2 mmol) and p-TsCl (3.8 g, 19.63 mmol) was added to a solution of 5 (3.2 g, 15.10 mmol) in CH2Cl2 (45 mL). After stirring overnight at room temperature, the reaction mixture was poured into water (110 mL) and extracted with CH2Cl2. The organic layer was washed with water, dried over MgSO4, filtered and concentrated in vacuo. The residue was dissolved in CH2Cl2 (70 mL) and then 2,6-lutidine (4.5 mL, 38.99 mmol) and TBSOTf (5.3 mL, 23.11 mmol) was added. After stirring at room temperature for 30 min, the solution was cooled to 0°C, satuated NaHCO3 was added and extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was chromatographed on silica gel (hexane) to give 5 as a colorless oil (4.29 g, 99%). 1H-NMR (CDCl3) δ 9.72 (dd, J = 1.3, 3.3 Hz, 1H), 3.99 (d, J = 2.6 Hz, 1H), 2.43 (m, 1H), 1.93 (m, 1H), 1.78 (m, 2H), 1.66 (m, 1H), 1.56 (m, 1H), 1.35 (m, 3H), 1.25 (m, 2H), 1.11 (m, 2H), 1.00 (d, J = 6.3 Hz, 3H), 0.96 (s, 3H), 0.89 (s, 9H), 0.01 (s, 3H), 0.002 (s, 3H).
(8S,20R)-De-A,B-8-(tert-Butyldimethylsilyl)oxy-20-(cyanomethyl)pregnane (7). To a solution of 6 (2.00 g, 4.17 mmol) in DMSO (30 mL) was added NaCN (270 mg, 5.42 mmol) and the resulting mixture was heated at 90°C for 1 h. The reaction mixture was poured into water (60 mL) and the organics were extracted with ethyl acetate. The extracts were washed with water and brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was chromatographed on silica gel (hexane) to give 7 (1.02 g, 72%) as a colorless oil. 1H-NMR (CDCl3) δ 4.01 (d, J = 2.6 Hz, 1H), 2.34 (dd, J = 4.0, 16.5 Hz, 1H), 2.23 (dd, J = 6.6, 16.5 Hz, 1H), 1.10~2.00 (m, 20H), 1.14 (d, J = 6.6 Hz, 3H), 0.93 (s, 3H), 0.89 (s, 9H), 0.02 (s, 3H), 0.00 (s, 3H).
(8S,20R)-De-A,B-8-(tert-Butyldimethylsilyl)oxy-20-(formylmethyl)pregnane (8). To a solution of DIBAH (1M in hexane) (6.1 mL, 6.1 mmol) in CH2Cl2 (20 mL) was added a solution of 7 (1.02 g, 3.05 mmol) in CH2Cl2 (25 mL) at 0°C with dropwise and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of saturated NH4Cl solution, and the resulting mixture was diluted with ether, and then stirred for another 30 min. The mixture was dried over MgSO4 and filtered through a pad of Celite, and the filtrates were concentrated in vacuo. The residue was chromatographed on silica gel (hexane) to give aldehyde 8 (595 mg, 58%) as a colorless oil. 1H-NMR (CDCl3) δ 9.75 (dd, J = 1.3, 3.3 Hz, 1H), 4.00 (d, J = 2.6 Hz, 1H), 2.46 (dd, J = 2.0, 13.5 Hz, 1H), 1.03~2.20 (m, 20H), 1.00 (d, J = 6.3 Hz, 3H), 0.96 (s, 3H), 0.89 (s, 9H), 0.01 (s, 3H), 0.002 (s, 3H).
(8S,20R)-De-A,B-8-(tert-Butyldimethylsilyl)oxy-20-[4-(2,5-dimethyl-5-isoxazolidinecarboxylic acid methyl ester)methyl]pregnane (11). To a mixture of 8 (595 mg, 2.66 mmol) in CH2Cl2 (8 mL) was added N-methyl hydroxylamine hydrochloride (184 mg, 3.19 mmol) and Et3N (1.0 mL), and the resulting mixture was stirred at room temperature for 3 h. To the reaction mixture was added saturated NaHCO3, and the mixture was extracted with CH2Cl2. The organic layer was dried over MgSO4 and concentrated in vacuo. The residue was dissolved in toluene (17 mL) and methyl methacrylate (10) (1.0 mL, 9.32 mmol) was added, and the resulting mixture was heated at 90°C for 2 h. The reaction mixture was concentrated in vacuo, and the residue was purified by HPLC using a PEGASIL Silica 60-5 column (ϕ 20x150 mm, Senshu Pak.) and elution with 15% ethyl acetate in hexane to give 11a (316.6 mg, 26%), 11b (343.6 mg, 28%), 11c (116.5 mg, 10%) and 11d (125.5 mg, 10%), respectively.
11a (23S, 25S): [α]24D = +112.4° (c 1.1, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.76 (s, 3H), 2.67 (s, 3H), 2.57 (br s, 1H), 2.46 (dd, J = 3.0, 13.1 Hz, 1H), 2.24 (dd, J = 7.0, 13.1 Hz, 1H), 1.94 (br d, J = 13.1 Hz, 1H), 1.79 (m, 2H), 1.65 (m, 2H), 1.54 (m, 1H), 1.47 (s, 3H), 1.39 ( m, 4H), 1.21 (m, 2H), 1.11 (m, 2H), 1.01 (m, 1H), 0.92 (d, J = 8.2 Hz, 3H), 0.91 (s, 3H), 0.88 (s, 6H), -0.01 (d, J = 7.0 Hz, 9H); 13C-NMR (CDCl3) δ 175.67, 77.25, 76.99, 76.74, 69.36, 66.95, 57.37, 53.03, 52.57, 45.87, 43.27, 42.17, 40.73, 38.06, 34.35, 34.03, 27.61, 25.77, 24.37, 23.01, 18.85, 17.99, 17.61, 13.74, -4.82, -5.21; IR (neat) 3318, 2952, 2856, 1738 cm-1; m/z 468 (M+H)+.
11b (23R, 25R): [α]24D = -26.7° (c 1.0, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.75 (s, 3H), 2.68 (s, 3H), 2.57 (dd, J = 9.0, 12.2 Hz, 1H), 2.51 (br s, 1H), 2.30 (m, 1H), 1.94 (br d, J = 12.2 Hz, 1H), 1.80 (m, 2H), 1.64 (m, 2H), 1.52 (m, 1H), 1.47 (s, 3H), 1.33 ( m, 3H), 1.23 (m, 3H), 1.13 (m, 2H), 1.00 (m, 1H), 0.92 (d, J = 6.0 Hz, 3H), 0.90 (s, 3H), 0.87 (s, 6H), -0.01 (d, J = 7.1 Hz, 9H); 13C-NMR (CDCl3) δ 175.69, 69.35, 68.00, 57.42, 52.97, 52.54, 47.64, 43.93, 42.22, 40.66, 38.83, 34.52, 34.34, 27.56, 25.77, 24.29, 23.02, 19.60, 17.99, 17.60, 13.65, -4,83, -5.21; IR (neat) 3287, 2951, 2857, 1738 cm-1; m/z 468 (M+H)+.
11c (23S, 25R): [α]24D = +62.5° (c 1.0, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.76 (s, 3H), 2.85 (dd, J = 6.1, 13.1 Hz, 1H), 2.70 (s, 3H), 2.00 (m, 1H), 1.86 (m, 1H), 1.66 (br d, J = 12.2 Hz, 1H), 1.57-1.42 (m, 1H), 1.52 (s, 3H), 1.37-0.99 (m, 12H), 0.91 (s, 3H), 0.89 ( d, J = 16.3 Hz, 3H), 0.88 (s, 6H), -0.01 (d, J = 7.2 Hz, 9H); 13C-NMR (CDCl3) δ 174.47, 77.24, 76.99, 76.74, 69.36, 66.78, 57.31, 53.02, 52.50, 46.82, 43,84, 42.18, 40.69, 38.38, 37.60, 34.34, 34.14, 29.66, 27.66, 25.77, 24.92, 24.83, 23.00, 22.62, 19.05, 18.90, 18.00, 17.60, 14.08, 13.72, -4.81, -5.21; IR (neat) 3790, 2951, 2856, 2777, 1739 cm-1; m/z 468 (M+H)+.
11d (23R, 25S): [α]24D = +16.9 (c 1.1, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.76 (s, 3H), 2.91 (dd, J = 6.1, 12.1 Hz, 1H), 1.95-1.91 (m, 2H), 1.81-1.76 (m, 2H), 1.70-1.64 (m, 2H), 1.59-1.47 (m, 2H), 1.51 (s, 3H), 1.37-1.20 (m, 6H), 1.12-0.97 (m, 3H), 0.92 (d, J = 9.2 Hz, 3H), 0.91 (s, 3H), 0.88 (s, 6H), -0.01 (d, J = 7.1 Hz, 9H); 13C-NMR (CDCl3) δ 174.84, 77.25, 76.99, 76.74, 69.38, 67.03, 57.42, 53.01, 52.48, 47.77, 44.88, 42.24, 40.68, 39.26, 34.37, 34.06, 31.56, 29.67, 27.51, 25.78, 24.86, 23.03, 22.62, 19.39, 18.00, 17.61, 14.08, 13.68, -4.81, -5.19; IR (neat) 3307, 2951, 2856, 2360, 1738 cm-1; m/z 468 (M+H)+.
(8S,20R,23S,25S)-De-A,B-8,25-Dihydroxy-cholestane-26,23-N-methyl lactam (12). To a solution of 11a (316.6 mg, 0.68 mmol) in MeOH (25 mL) was added a catalytic amount of p-TsOH and the resulting mixture was stirred for 12 h. The reaction mixture was diluted with ethyl acetate and the saturated NaHCO3 was added. The resulting mixture was extracted with ethyl acetate and the organics was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was chromatographed on silica gel (5:1 hexane:ethyl acetate) to give the alcohol (218.7 mg, 91%) as a colorless oil. [α]24D = +145.9 (c 1.0, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.68 (s, 3H), 2.58 (s, 3H), 2.49 (br s, 1H), 2.38 (dd, J = 9.0, 13.0 Hz, 1H), 2.15 (dd, J = 8.2, 13.0 Hz, 1H), 1.90 (br d, J = 13.0 Hz, 1H), 1.80-1.69 (m, 3H), 1.54-0.93 (m, 11H), 1.39 (s, 3H), 0.84 (d, J = 6.3 Hz, 3H), 0.84 (s, 3H); 13C-NMR (CDCl3) δ 175.71, 69.26, 66.90, 57.24, 52.60, 52.59, 45.83, 43.28, 41.91, 40.41, 38.06, 34.03, 33.54, 27.45, 24.36, 22.47, 18.77, 17.38, 13.51; IR (neat) 3526, 2948, 2867, 2360, 1737 cm-1; m/z 354 (M+H)+. A mixture of the alcohol (218.7 mg, 0.62 mmol) and 10% Pd/C (100 mg) in EtOH (15 mL) was stirred under H2 atmosphere for 12 h. The reaction mixture was filtered through a pad of Celite and the filtrates were concentrated in vacuo to give 12 (180 mg, 90%) as a colorless oil. [α]24D = +145.9° (c 1.0, CHCl3); 1H-NMR (CDCl3) δ 3.98 (br s, 1H), 3.68 (s, 3H), 2.58 (s, 3H), 2.49 (br s, 1H), 2.38 (dd, J = 9.0, 13.1 Hz, 1H), 2.15 (dd, J = 8.0, 13.1 Hz, 1H), 1.90 (br d, J = 13.1 Hz, 1H), 1.80-1.69 (m, 3H), 1.54-0.93 (m, 11H), 1.39 (s, 3H), 0.84 (d, J = 6.0 Hz, 3H), 0.84 (s, 3H); 13C-NMR (CDCl3) δ 175.71, 69.26, 66.90, 57.24, 52.60, 52.59, 45.83, 43.28, 41.91, 40.41, 38.06, 34.03, 33.54, 27.45, 24.36, 22.47, 18.77, 17.38, 13.51; IR (neat) 3381, 2936, 2867, 2242, 1680 cm-1; m/z 324 (M+H)+.
(20R,23S,25S)-De-A,B-25-Trimethylsilylhydroxy-cholestane-8-one-26,23-N-methyl lactam (13). To a solution of 12 (212 mg, 0.66 mmol) in CH2Cl2 (5 mL) was added tetra-n-propylammonium perruthenate (12 mg, 0.66 mmol), 4-methylmorpholine N-oxide (310 mg, 2.64 mmol) and molecular sieves 4A powder, and the whole mixture was stirred for 2 h at room temperature. The reaction mixture was loaded on the silica gel column chromatography (1:2 hexane - ethyl acetate) to give the ketone (219 mg, quant. yield) as a colorless oil. A mixture of the ketone (95 mg, 0.3 mmol) and TMS-imidazole (0.22 mL, 1.5 mmol) in CH2Cl2 (5 mL) was stirred for 12 h at room temperature. The reaction mixture was cooled at 0°C and water was added to the mixture. The organic layer was extracted with CH2Cl2, and the extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was chromatographed on silica gel (2:1 hexane - ethyl acetate) to give 13 (107 mg, 91%) as a colorless oil. [α]24D = +36.0° (c 1.0, CHCl3); 1H-NMR (CDCl3) δ 3.55(m, 1H), 2.78 (s, 3H), 2.46 (dd, J = 8.0, 12.0 Hz, 1H), 2.32-1.20 (m, 19H), 1.02 (d, J = 6.1 Hz, 3H), 0.66 (s, 3H), 0.11 (s, 9H); 13C-NMR (CDCl3) δ 211.49, 174.56, 76.24, 61.95, 57.11, 53.90, 49.81, 43.41, 40.89, 39.74, 39.03, 32.47, 27.94, 27.70, 25.54, 23.96, 19.04, 18.68, 12.53, 1.75; IR (neat) 3035, 2957, 2360, 1705 cm-1; m/z 394 (M+H)+.
(7E)-(20R,23S,25S)-De-A,B-8-Bromomethylidene-25-trimethylsilylhydroxy-cholestane-26,23-N-methyl lactam (14). To an emulsion of bromomethyltriphenylphosphonium bromide (327 mg, 0.75 mmol) in toluene (2 mL) was added lithium hexamethyldisilazane (1 M in THF) (0.74 mL, 0.74 mmol) at -78°C and the mixture was warmed up to 0°C and stirred for 10 min. After cooling to -78°C, to the resulting solution was added 13 (60.1 mg, 0.15 mmol) in toluene (1 mL) with dropwise, and the mixture was stirred at the same temperature for 2 h. To the reaction mixture was added saturated NH4Cl and warmed up to room temperature. The resulting mixture was extracted with ethyl acetate and the organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane) to give 14 (8.4 mg, 12%) and 13 (32 mg, 52%) was recovered. [α]24D = +79.47° (c 0.9, CHCl3); 1H-NMR (CDCl3) δ 5.56 (s, 1H), 3.45 (m, 1H), 2.80 (m, 1H), 2.68 (s, 3H), 2.19 (dd, J = 6.0, 12.2 Hz, 1H), 1.94-1.10 (m, 18H), 0.90 (d, J = 6.0 Hz, 3H), 0.49 (s, 3H), 0.02 (s, 9H); 13C-NMR (CDCl3) δ 174.79, 144.73, 97.70, 56.27, 55.89, 53.93, 45.52, 43.38, 39.90, 39.80, 32.92, 30.97, 29.69, 28.01, 27.70, 25.58, 22.48, 21.98, 18.76, 11.87; IR (neat) 3734, 2952, 1700, 1635, 1507 cm-1; m/z 470, 472 (M+H)+.
(2S)-2,4-Diol butanol anisilidene (17). To a solution of boranemethyl sulfide complex (100 mL, 1 mol) in THF (200 mL) was added the solution of (S)-malic acid (15) (50 g, 373 mmol) in THF (400 mL) at 0°C with dropwise over 3 h, and the resulting mixture was stirred for another 12 h at room temperature. The reaction mixture was re-cooled to 0°C and MeOH (400 mL) was added. The organics were evaporated in vacuo and the residue was dissolved in MeOH (300 mL) and evaporated in vacuo. The same sequences (addition of MeOH and concentration of organics) were repeated three times. The residue was dissolved in MeOH (300 mL) and p-TsOH (200 mg) was added, and the mixture was evaporated in vacuo to give 16 (50.63 g, 99%) as a colorless oil. To a solution of 16 (15 g, 141 mmol) in MeOH (240 mL) was added CSA (3.3 g, 14 mmol) and the mixture was heated at 90°C for 1 h. The resulting mixture was cooled to room temperature, and the organics were evaporated in vacuo. The residue was dissolved in CH2Cl2 and 4-methoxybenzaldehyde dimethylacetal (30 mL, 170 mmol) was added to the solution, and the resulting mixture was stirred for 6 h. To the reaction mixture was added saturated NaHCO3, and the organic layer was extracted with ethyl acetate and washed with brine. The aqueous layer was extracted twice with ethyl acetate. The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (2:1 hexane - ethyl acetate) to give 17 (18.23 g, 26%) as a pale yellow oil.
(3S)-3,5-Diol-1-pentene anisilidene (18). To a solution of (COCl)2 (6.0 mL, 67.4 mmol) in CH2Cl2 ( 50 mL) was added DMSO (12.0 mL, 168.5 mmol) dropwise at –78°C and the mixture was stirred for 15 min. To the resulting mixture was added 17 (8.23 g, 33.7 mmol) in CH2Cl2 (100 mL) and the mixture was stirred for 40 min. Et3N (470 mL, 337 mmol) was added and whole mixture was warmed to room temperature, and then stirred for another 30 min. Water (20 mL) was added and the organic layer was extracted with ethyl acetate. The extracts were washed with brine, dried over MgSO4, filtered and evaporated in vacuo. The residue in THF solution was added to a solution of methylphosphoniumylide in THF prepared from methylphosphonium bromide (16.6 g, 46.5 mmol) and n-BuLi (1.5 M in hexane, 30 mL, 45 mmol) in THF (250 mL) at 0°C and the whole mixture was stirred at 0°C for 30 min and at room temperature for another 1 h. After cooling to 0°C, water was added, and the organics were extracted with ethyl acetate. The extracts were washed with brine, dried over MgSO4, filtered and evaporated in vacuo. The residue was chromatographed on silica gel (10:1 hexane - ethyl acetate) to give 18 (2.77 g, 38%) as a colorless oil. 1H-NMR (CDCl3) 7.44 (d, J = 10.0 Hz, 2H), 6.88 (d, J = 10.0 Hz, 2H), 5.99 (m, 1H), 5.58 (s, 1H), 5.38 (dd, J = 13.1, 1.0 Hz, 1H), 5.19 (dd, J = 13.1, 1.0 Hz, 1H), 4.36 (dd, J = 12.2, 6.3 Hz, 1H), 4.28 (m, 1H), 4.00 (m, 1H), 3.80 (s, 3H), 1.92 (m, 1H), 1.60 (m, 1H).
(3S)-3-Methoxyphenylmethylyloxy-5-hydroxy-1-pentene (19). To a solution of 18 (2.77 g, 12.6 mmol) in CH2Cl2 (130 mL) was added DIBAH (0.95 M in hexane, 66 mL, 63 mmol) dropwise at -20°C and the mixture was stirred for 2 h. 2-Propanol was added, then water and silica gel. The resulting slurry was diluted with ethyl acetate and stirred at room temperature for another 1 h. The slurry was filtered through a pad of Celite and the filtrates were concentrated in vacuo. The residue was purified by silica gel column chromatography (10:1 hexane - ethyl acetate) to give 19 (1.15 g, 41%) as a colorless oil. 1H-NMR (CDCl3) δ 7.14 (d, J = 9.3 Hz, 2H), 6.77 (d, J = 9.3 Hz, 2H), 5.68 (m, 1H), 5.17 (m, 2H), 4.46 (d, J = 12.2 Hz, 2H), 4.20 (d, J = 12.2 Hz, 2H), 3,90 (m, 1H), 3.70 (s, 3H), 3.65 (m, 1H), 1.75 (m, 1H), 1.67 (m, 1H).
(3S)-3-[(Methoxyphenylmethylyl)oxyl-5-formyl-1-pentene (20). A mixture of 19 (1.15 g, 5.17 mmol) , NaHCO3 (1.7 g, 20.68 mmol) and Dess-Martin periodinane (4.4 g, 10.34 mmol) in CH2Cl2 (45 mL) was stirred for 1 h. The reaction mixture was directly loaded on a silica gel column (10:1 hexane - ethyl acetate) and purified to give 20 (990 mg, 89%) as a colorless oil. 1H-NMR (CDCl3) δ 9.48 (s, 1H), 7.15 (d, J = 10.1 Hz, 2H), 6.77 (d, J = 10.1 Hz, 2H), 5.50 (m, 1H), 5.04 (d, J = 17.4 Hz, 1H), 4.97 (d, J = 10.1 Hz, 1H), 4.41 (d, J = 13.2 Hz, 1H), 4.19 (d, J = 13.2 Hz, 1H), 4.00 (m, 1H), 2.35 (m, 1H), 2.2 (m, 1H).
(3S)-3-[(Methoxyphenylmethylyl)oxyl]-5-hydroxy-1-octen-7-yne (21). To a mixture of Mg (220 mg, 2.25 mmol) and HgCl2 (25 mg, 0.09 mmol) in ether (40 mL) was added propargyl bromide (0.75 mL, 9.9 mmol) at room temperature until the Mg was dissolved. The mixture was cooled to 0°C, and 20 (495 mg, 2.25 mmol) in ether (25 mL) was added dropwise. After being stirred for 15 min, saturated NH4Cl solution was added to the reaction mixture. The organics were extracted with ethyl acetate and washed with brine. The extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (8:1 hexane - ethyl acetate) to give 21 (393.1 mg, 67%) as a colorless oil.
(3S,5R)-3,5-Dihydroxy-1-octen-7-yne (23a). To a solution of 21 (741.5 mg, 2.8 mmol) in CH2Cl2 (50 mL) was added DDQ (953 mg, 4.2 mmol) at room temperature. After being stirred for 1 h, the reaction mixture was quenched by the addition of saturated NaHCO3. The mixture was extracted with CH2Cl2, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (8:1 hexane - ethyl acetate) to give 23a (98.7 mg, 25%) and 22b (189 mg, 26%) as colorless oils. 23a: 1H-NMR (CDCl3) δ 5.94 (m, 1H), 5.31 (d, J = 10.2 Hz, 2H), 5.17 (d, J = 10.2 Hz, 2H), 4.49 (m, 1H), 4.41 (m, 1H), 4.11 (m, 1H), 4.05 (m, 1H), 2.41 (m, 2H), 2.16 (m, 1H), 1.86 (m, 1H), 1.73 (m, 1H).
(3S,5R)-3,5-Di[(t-butyldimethylsilyl)oxyl]-1-octen-7-yne (24). To a solution of 23a (98.7 mg, 0.70 mmol) in CH2Cl2 (7 mL) was added 2,6-lutidine (0.25 mL, 2.1 mmol) and TBSOTf (0.4 mL, 1.75 mmol) at 0°C and the mixture was stirred for 30 min at room temperature. The reaction mixture was re-cooled to 0°C, and saturated NaHCO3 was added. The organics were extracted with CH2Cl2 and the extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was chromatographed on silica gel (hexane) to give 24 (248.2 mg, 96%) as a colorless oil. [α]24D = -5.19° (c 0.7, CHCl3); 1H-NMR (CDCl3) δ 5.57 (m, 1H), 5.04 (d, J = 17.4 Hz, 1H), 4.94 (d, J =10.1 Hz, 1H), 4.11 (m, 1H), 3.82 (m, 1H), 2.25 (m, 2H), 1.87 (m, 1H), 1.81-1.53 (m, 2H), 1.44 (s, 1H), 0.78 (m, 18H), 0.03 (m, 12H); 13C-NMR (CDCl3) δ 141.90, 114.25, 81.47, 71.65, 70.07, 68.13, 45.89, 27.98, 25.92, 25.85, -3.76, -4.15, -4.45, -4.58; IR (neat) 3315, 2956, 2930, 2888, 2858, 1749, 1624, 1508 cm-1; m/z 369 (M+H)+.
(23S,25S)-1α,3-Di[(t-butyldimethylsilyl)oxyl]-25-(trimethylsilyl)oxyl vitamin D3 26,23-N-methyl lactam (25). A mixture of Pd2(dba)3CHCl3 (2 mg, 0.002 mmol), PPh3 (5 mg, 0.19 mmol) and Et3N (0.8 mL) in toluene (0.8 mL) was stirred for 10 min at room temperature. To the resulting mixture was added 14 (13.8 mg, 0.03 mmol) and 24 (8 mg, 0.02 mmol) in toluene (0.8 mL) dropwise, and the whole mixture was heated at 110°C for 4 h. The reaction mixture was diluted with hexane and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane) to give 25 (9.4 mg, 62%) as a colorless oil. Ketone 14 (4.7 mg, 33%) and enyne 24 were recovered (2.7 mg, 37%).
(23S,25S)-1α,25-Dihydroxy vitamin D3-26,23-N-methyl lactam (3a). A mixture of 25 (4.7 mg, 0.0062 mmol) and HF pyridine (1 mL) in THF (2 mL) at 0°C was stirred for 6 h. The reaction mixture was quenched with saturated NaHCO3 and the organics were extracted with ethyl acetate. The extracts were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (9:1 CHCl3-MeOH) to give 3a as a pale yellow oil (1.5 mg, 53%). 1H-NMR (CDCl3) δ 6.37 (d, J = 11.5 Hz, 1H), 6.02 (d, J = 11.1 Hz, 1H), 5.33 (s, 1H), 4.99 (s, 1H), 4.43 (br s, 1H), 4.23 (br s, 1H), 2.82 (s, 3H), 2.61-0.70 (m, 33H), 0.99 (d, J = 6.4 Hz, 3H), 0.58 (s, 3H); m/z 458(M+H)+.
References
- Bouillon, R.; Okamura, W. H.; Norman, A. W. Structure-function relationships in the Vitamin D endocrine system. Endocrine Rev. 1995, 16, 200–257. [Google Scholar]
- Ishizuka, S.; Norman, A. W. The difference of biologial activity among four diastereoisomers of 1α,25-dihydroxycholecalciferol-26,23-lactone. J. Steroid. Biochem. 1986, 25, 505–510. [Google Scholar] [CrossRef]
- Iwasaki, H.; Hosotani, R.; Miyamoto, Y.; Nakano, Y. Stereoselective synthesis and structural establishment of (25S)-24,24-difluoro-1α,25,26-trihydroxyvitamin D3, a major metabolite of 24,24-difluoro-1α,25-dihydroxyvitamin D3. Tetrahedron. 1998, 54, 14705–14724. [Google Scholar] [CrossRef]
- Wovkulich, P. M.; Barcelos, F.; Batcho, A. D.; Sereno, J. F.; Baggiolini, E. G.; Hennessy, B. M.; Uskokovic, M. R. Stereoselective total synthesis of 1α,25S,26-trihydroxycholeccalciferol. Tetrahedron 1984, 40, 2283–2296. [Google Scholar] [CrossRef]
- Griffith, W. P.; Ley, S. V.; Whitcombe, G. P.; White, A. D. Preparation and use of tetra-n-butylammonium per-ruthenate (TBAP reagent) and tetra-n-propylammonium per-ruthenate (TPAP reagent) as new catalytic oxidants for alcohols. J. Chem. Soc., Chem. Commun. 1987, 1625–1627. [Google Scholar]
- Trost, B. M.; Dumas, J.; Villa, M. New strategies for the synthesis of vitamin D metabolites via Pd- catalyzed reactions. J. Am. Chem. Soc. 1992, 114, 9836–9845. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Sakurai, K.; Saijo, K.; Takano, K. Enantioselective synthesis of (6S,7S,9R,10R)-6,9-epoxynonadec-18-ene-7,10-diol, a marine natural product. Tetrahedron Lett. 1985, 26, 1333–1336. [Google Scholar] [CrossRef]
- Dess, D. B.; Martin, J. C. A useful 12-I-5 triacetoxyperiodinane (the Dess-Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Ikeda, M.; Takahashi, K.; Dan, A.; Koyama, K.; Kubota, K.; Tanaka, T.; Hayashi, M. Synthesis and biological evaluations of A-ring isomers of 26,26,26,27,27,27-hexafluoro-1,25-dihydroxyvitamin D3. Bioorg. Med. Chem. 2000, 8, 2157–2166. [Google Scholar]
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Kato, Y.; Hashimoto, Y.; Nagasawa, K. Novel Heteroatom-containing Vitamin D3 Analogs: Efficient Synthesis of 1α,25-Dihydroxyvitamin D3-26,23-lactam. Molecules 2003, 8, 488-499. https://doi.org/10.3390/80600488
AMA Style
Kato Y, Hashimoto Y, Nagasawa K. Novel Heteroatom-containing Vitamin D3 Analogs: Efficient Synthesis of 1α,25-Dihydroxyvitamin D3-26,23-lactam. Molecules. 2003; 8(6):488-499. https://doi.org/10.3390/80600488
Chicago/Turabian StyleKato, Yuko, Yuichi Hashimoto, and Kazuo Nagasawa. 2003. "Novel Heteroatom-containing Vitamin D3 Analogs: Efficient Synthesis of 1α,25-Dihydroxyvitamin D3-26,23-lactam" Molecules 8, no. 6: 488-499. https://doi.org/10.3390/80600488