Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents

1
University of Applied Sciences Basel, Gründenstrasse 40, CH-4132 Muttenz, Switzerland
2
Actelion Pharmaceuticals Ltd, Gewerbestrasse 16, CH-4123 Allschwil, Switzerland
*
Author to whom correspondence should be addressed.
Molecules 2003, 8(7), 556-564; https://doi.org/10.3390/80700556
Submission received: 16 June 2003
/
Revised: 25 June 2003
/
Accepted: 25 June 2003
/
Published: 15 July 2003
Abstract
:A new series of plasmepsin II (PM II) inhibitors has been prepared based on 4-aminopiperidine-tert-butyl-carbamate (1). These compounds might be useful as antimalarial drugs acting via a new mechanism, and therefore be less susceptible to parasite resistance now often observed with current antimalarial therapies. Some of the final compounds prepared exhibited encouraging inhibitory activity towards PM II.
Introduction
The World Health Organization (WHO) estimates that more than 280 million people are infected with malaria. This situation worsens because drug resistant strains of the malaria parasite are spreading quickly. Among four different strains, Plasmodium falciparum causes the largest number of lethal infections and often shows substantial resistance against Chloroquin, still the most important drug used to treat malaria infections.
Plasmodia are transmitted by Anopheles gambia mosquitoes, which are developing greater pesticide resistance. Climate changes, changing agricultural habits and increased mobility are assumed to be responsible for the spread of the mosquito and the disease into formerly unaffected regions. Political conflicts in regions affected by malaria have made disease control difficult. Thus, possible new drug targets are urgently needed which allow for the development of new medicines acting via different mechanisms in order to avoid fast development of resistance [1].
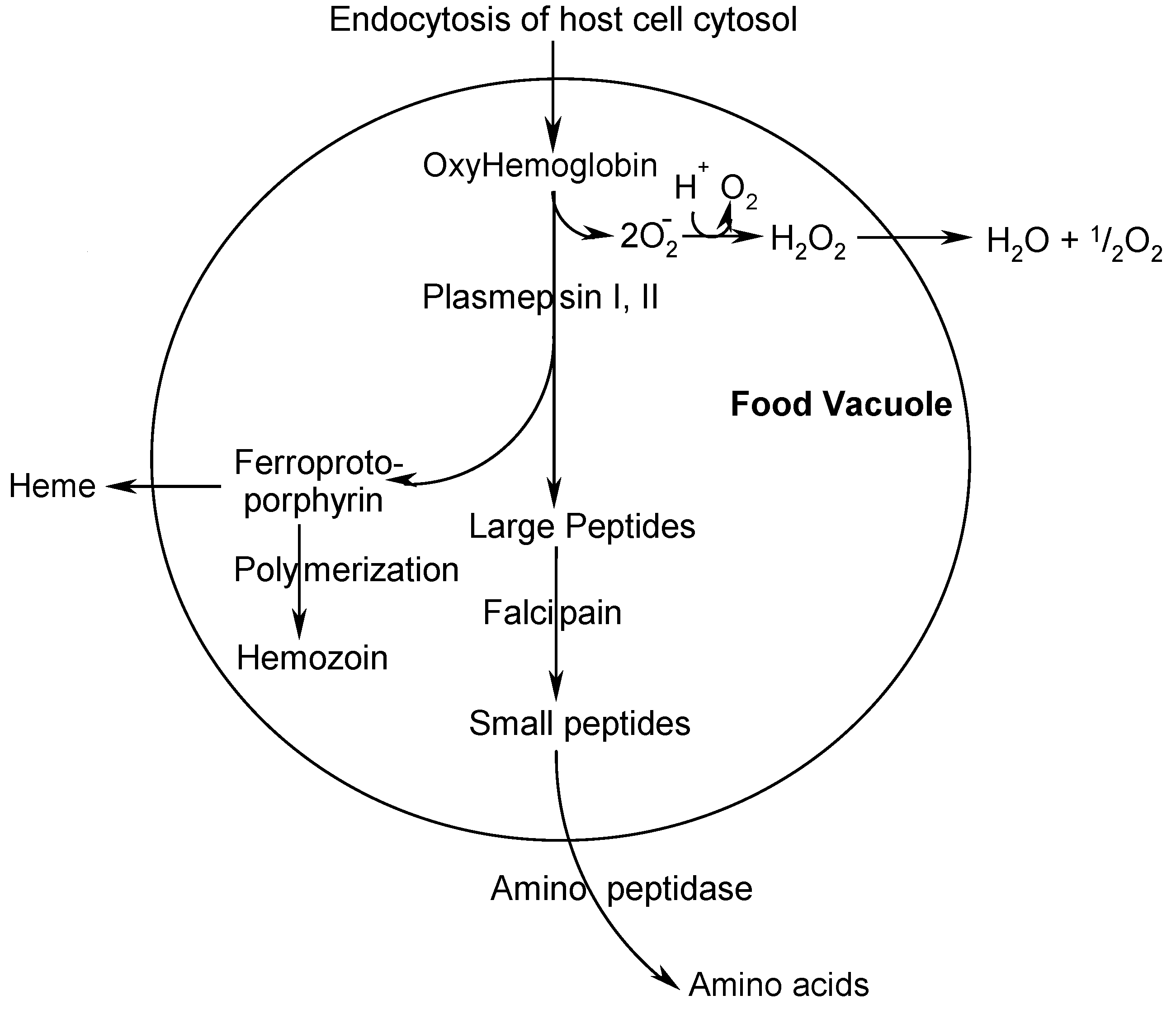
Among the various targets, the hemoglobin degradation pathway - involving several parasite specific enzymes like the plasmepsins [6] and the falcipains [2] - is of major interest. One possibility to strike against the parasite is to inhibit an enzyme within this pathway [2], interrupting the nutrition source and therefore eliminating the parasite by starvation.
Scheme 1.

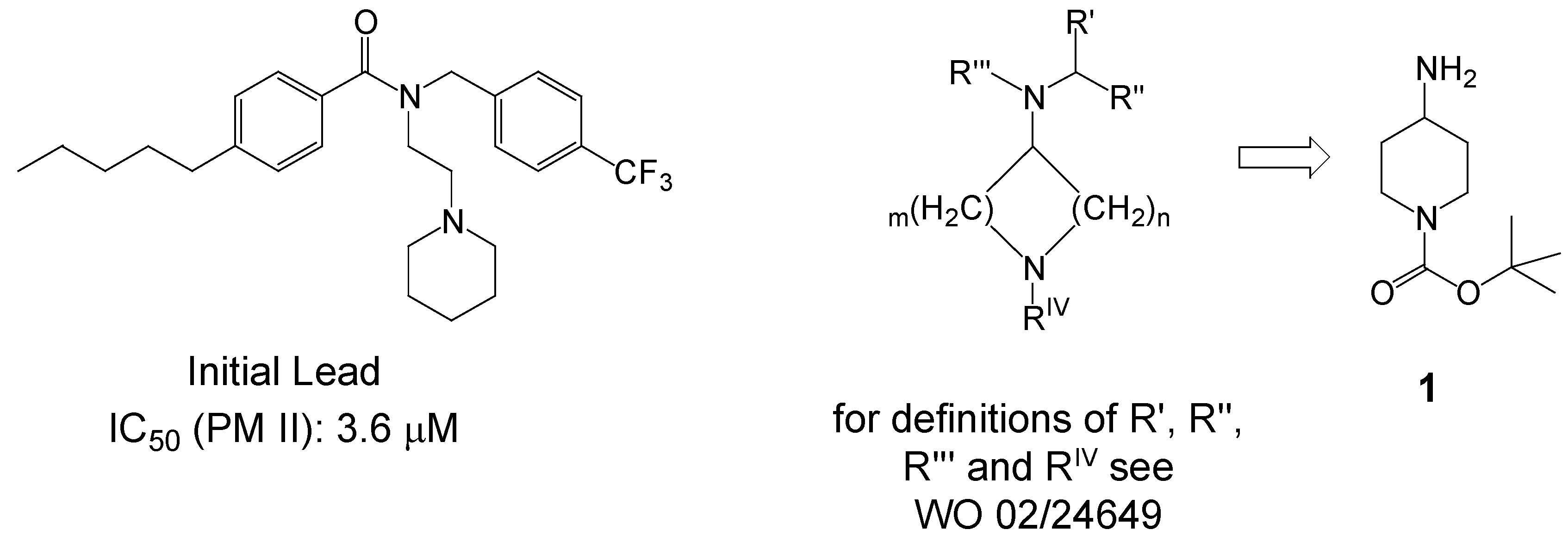
Scheme 1 presents a simplified overview of processes in the food vacuole and shows many interesting targets for drug development [3,4,5]. PM II has been chosen because its inhibition follows a completely new and promising pathway of fighting against the malaria parasite having a good potential of being devoid of fast emerging resistance. This target fits well within our core competencies in the field of aspartic protease inhibition as a therapeutic strategy [6], which has been proven especially valid in the field of HIV-protease inhibition to treat AIDS. The lead compounds depicted below (Scheme 2) emerged from a high throughput screening (HTS) of a commercial library containing 50,000 compounds for PM II inhibitory activity.
One of the preferred lead structures found in the HTS effort considered as being suitable for optimization as PM II inhibitors is depicted in Scheme 2 exhibiting an IC50 on PM II of 3.6 μM. By variation of different parameters like ring size, functional groups of substituents or chain length of alkyl groups, the preferred key intermediate identified for further optimization cycles was 4-amino-piperidine-tert-butylcarbamate (1) [7].
Scheme 2.

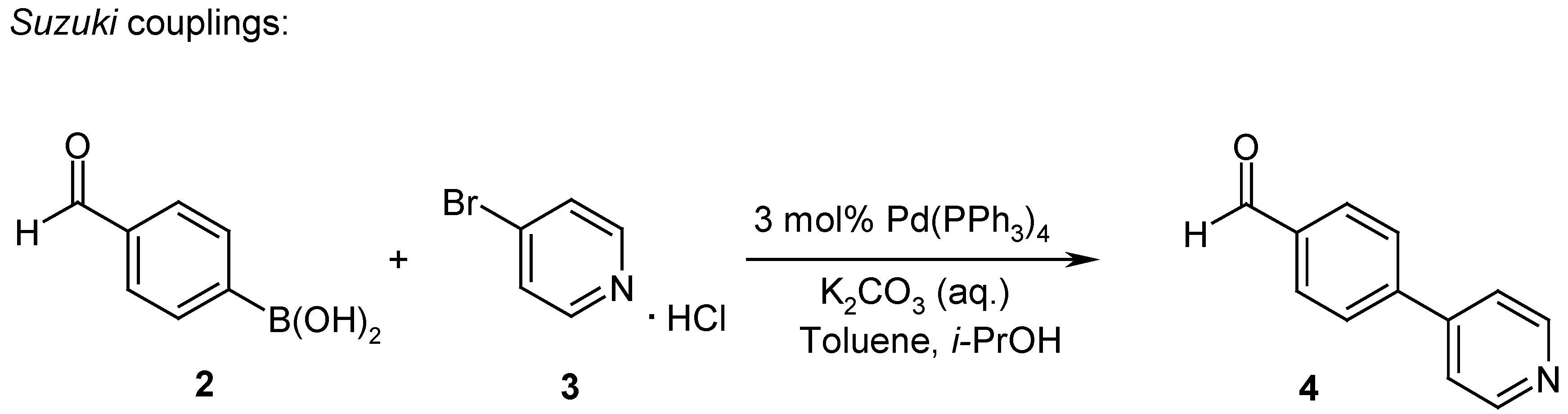
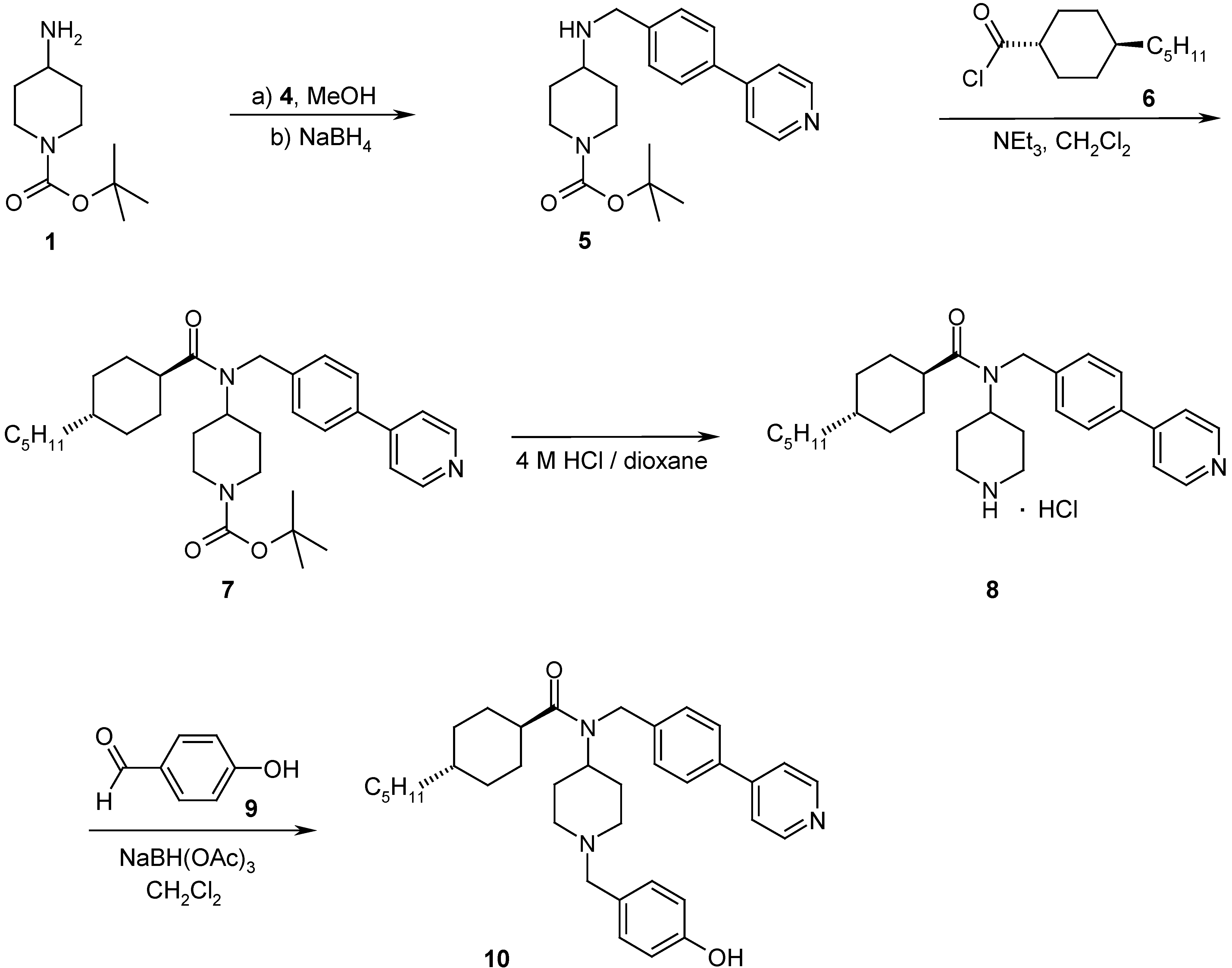
Part of the work presented here, was undertaken to elaborate the Structure-Activity-Relationships (SAR) with respect to variations of RII and RIV (Scheme 2, RI=H; for RIII either the trans-4-pentyl-cyclohexanoyl- or the 4-pentyl[2.2.2]bicyclooctanoyl-unit was introduced as a replacement of the 4-pentyl-benzoyl-group for which results are published in [6]). Therefore the synthesis of biaryl aldehydes 4 by Suzuki coupling reactions [8] (Scheme 3) was investigated and optimized. Aldehydes 4 were then coupled in a reductive amination reaction onto N-Boc-4-aminopiperidine (1) [7,9]. The subsequent acylation of amine 5 with suitable carboxylic acids or carboxylic acid chlorides (e.g. 6) under standard reaction conditions resulted in the amides 7. Cleavage of the Boc-protecting group with 4 M HCl in dioxane gave the precursors 8. Final reductive amination with a diverse set of aldehydes (e.g. 9) following standard literature procedures in a parallel chemistry set-up resulted in the target inhibitors 10 which were tested for inhibition of plasmepsin II in a FRET-assay [10]. In order to assess the specificity and the selectivity of the PM II inhibitors (e.g. 10), the compounds were tested in FRET-assays for the inhibition of other aspartic proteases like plasmepsin IV, cathepsin D or cathepsin E. For the whole set of final compounds depicted in Table 1 and Table 2, substantially lower or no inhibition of these aspartic proteases was detected.
The main goal of this investigation was to find out what types of biaryl units in combination with either the trans-4-pentylcyclohexanoyl- or the 4-pentyl[2.2.2]bicyclooctanoyl-unit in position RIII led to the most active PM II inhibitors (e.g. 10; Scheme 4). In order to confirm the superior influence of a biaryl moiety on the inhibitory activity, a limited number of compounds bearing a 4-alkyl- or 4-alkoxy-substituted benzyl group were also prepared (see Table 1 and Table 2).
Scheme 3.

Scheme 4.
Representative synthetic pathways for the preparation of 4-amino-piperidine based PMII inhibitors
Scheme 4.
Representative synthetic pathways for the preparation of 4-amino-piperidine based PMII inhibitors

Results and Discussion
Table 1 and Table 2 summarize the results for the two series of PM II inhibitors prepared according to the pathway depicted in Scheme 4. Table 1 shows compounds bearing the trans-4-pentyl-cyclohexanoyl unit as RIII. With respect to R1 the differences in activity between the compounds containing a 4-pyridyl moiety and the compounds containing a 3-pyridyl moiety are in the range of a factor of 2, with no clear preference for either pyridyl moiety. Replacement of the pyridyl units by an ethoxy group results in a 6- to 10-fold loss of activity (with the exception of the compounds given in row 6, Table 1, with R2 = 2-imidazolyl, where already the pyridyl substituted compound exhibits low activity). With respect to R2, the small lipophilic 2-methyl-propyl unit leads to more active compounds than the introduction of larger, heteroatom containing (e.g. hydroxy-phenyls (rows 2, 3, Table 1) or pyridyls (rows 4, 5, Table 1) or highly hydrophilic units like 2-imidazolyl, leading to an almost complete loss of activity in this series.



Table 2 gives the results for the PM II inhibitors bearing the 4-pentyl[2.2.2]bicyclooctanoyl moiety in position RIII. The compounds possessing a 4-pyridyl group in position R1 are generally more active than the compounds having an isopropyl unit in this position. This reflects the same SAR tendencies already elaborated for the first series of inhibitors given in Table 1. In the second group of inhibitors (Table 2) the introduction of a 2-imidazolyl unit in position R2 is much less detrimental for the inhibitory activity (col. 1, row 4, Table 2). But again the small lipophilic 2-methyl-propyl group leads to the most active derivatives. The introduction of the 4-hydroxy-phenyl (row 2, Table 2) or the 4-pyridyl (row 3, Table 2) unit results in derivatives exhibiting comparable activities as analogous compounds from Table 1. The influence on changes in activity by moving from trans-4-pentyl-cyclohexanoyl as RIII to 4-pentyl[2.2.2]bicyclooctanoyl as RIII seems to be weak. Both substituents lead to the same SAR within the respective series of PM II inhibitors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
The most active compounds prepared within these optimization efforts exhibit potencies which are a 100-fold higher than the activity of the initial lead compound depicted in Scheme 2. It has been shown that 4-aminopiperidine might well serve as a template to develop non-peptidic inhibitors of PM II allowing for the use of a parallel chemistry setup for the fast preparation of small focused libraries. In future work the potency of the PM II inhibitors series described needs to be further improved. In addition the physicochemical properties of the inhibitors (e.g. clogP, polar surface area or molecular weight) have to be optimized towards more drug-like values. Investigations are ongoing and results will be published in due course. The rather poor physicochemical properties of the final compounds depicted in Table 1 and Table 2 could raise questions about a non-specific interaction of the inhibitors with the enzyme. According to preliminary results obtained from X-ray analysis there is strong evidence for a specific interaction of the inhibitors with the active site of the enzyme. This fact confirms the results obtained in the selectivity screening discussed above.
Experimental
General
1H-NMR spectra were recorded at 300 MHz using a Varian Oxford NMR 300 and chemical shifts are reported in δ values (ppm) using DMSO-d6 as internal standard (unless otherwise specified). Purities of all compounds were over 95% and were determined by either 1H-NMR or LC-MS (Waters Micromass; ZMD-platform with ESI-probe with Alliance 2790 HT; Colum: 2x30mm, Gromsil ODS4, 3μm, 120A; Gradient: 0 – 100% acetonitrile in water, 6 min, with 0.05% formic acid, flow: 0.45mL/min). All solvents were stored over molecular sieves, except wet solvents used in Suzuki reactions. All reagents were used as received from commercial sources without further purification.
Selected Procedures [11]
4-Pyridin-4-yl-benzaldehyde (4)
To a solution of 3-bromopyridine hydrochloride (3) (2.92 g, 15 mmol, 1 eq) in toluene (30 mL) was added iso-propanol (30 mL), 4-formylphenylboronic acid (2) (2.48 g, 16.5 mmol, 1.1 eq) and 2 M aqueous K2CO3 (40 mL, 80 mmol, 5.3 eq). The solution was degassed with N2 for 15 minutes. After adding Pd(PPh3)4 (0.519 g, 0.45 mmol, 0.03 eq), the solution was refluxed for 7 h under argon. The reaction mixture was diluted with water and extracted three times with ethyl acetate. The combined organic layers were washed twice with brine, dried over MgSO4 and the solvent was evaporated. Recrystallization from ethyl acetate gave 2.13 g (77%) of 4-pyridin-4-yl-benzaldehyde (4) as yellow needles. LC: tr 2.41’; MS: 184 [M+H]+; 1H-NMR: 10.10 (s, 1H), 8.71 (d, 2H), 8.06 (s, 4H), 7.77 (d, 2H).
4-(4-Pyridin-4-yl-benzylamino)-piperidine-1-tert-butyl carbamate (5)
A solution of 4 (1.07 g, 5.33 mmol, 1 eq) and 1 (976 mg, 5.33 mmol, 1 eq) in methanol (30 mL) was refluxed for 4 h. After cooling to r.t., NaBH4 (222 mg, 5.87 mmol, 1.1 eq) was added carefully over 30 minutes. After 2 h of additional refluxing, the solution was carefully diluted with water and extracted three times with ethyl acetate. The combined organic layers were washed twice with brine, dried over MgSO4 and the solvent was evaporated to give 1.90 g (96%) of compound 5 as a yellow solid which could be used in further transformations without purification [7]. LC: tr 2.68’; MS: 368 [M+H]+; 1H-NMR: 8.63 (d, 2H), 7.82-7.41 (m, 6H), 3.89-3.74 (m, 3H), 3.79 (s, 1H), 3.08-1.73 (m, 5H) 1.40 (br s, 9H) 1.28-1.09 (m, 2H).
4-[(4-trans-Pentyl-cyclohexanecarbonyl)-(4-pyridin-4-yl-benzyl)-amino]-piperidine-1-tert-butyl carbamate (7)
To a solution of trans-4-pentylcyclohexanecarboxylic acid (485 mg, 2.45 mmol, 1 eq) in CH2Cl2 (20 mL) at 0°C was added oxalic acid chloride (0.35 mL, 4.14 mmol, 1.7 eq) and 3 drops of DMF. The mixture was stirred in an N2 atmosphere for 1 h and carefully evaporated (40°C, 200 mbar, argon at inlet) [12]. The obtained yellowish viscous acid chloride 6 was dissolved in an Ar atmosphere in CH2Cl2 (10 mL), followed by the addition of NEt3 (1.2 mL, 8.61 mmol, 3.5 eq) and 5 (900 mg, 2.45 mmol, 1 eq) dissolved in CH2Cl2 (10 mL). Stirring was continued at r.t. for 12 h. The mixture was diluted with ethyl acetate, washed once with saturated Na2CO3 solution and with brine, dried over MgSO4 and the solvent was evaporated. The crude material was purified by column chromatography (silica gel 60, DCM/Hex/MeOH 10:4:1) to give 623 mg (46%) of the amide 7 as white filaments [7]. LC: tr 5.85’; MS: 548 [M+H]+; 1H-NMR in CDCl3 (shows both amide rotamers with a ratio of ~1:1): 8.63-8.52 (m, 2H), 7.65-7.14 (m, 6H), 4.73-4.60 (m, 1H), 4.31, 4.24 (s, 2H, benzylic methylene of both rotamers), 4.19-3.92 (m, 2H), 2.77-0.56 (m, 36H).
4-trans-Pentyl-cyclohexanecarboxylic acid piperidin-4-yl-(4-pyridin-4-yl-benzyl)-amide hydrochloride (8)
A solution of compound 7 (617 mg, 1.13 mmol, 1 eq) in 4 M HCl / dioxane (10 mL, 40 mmol, 35.4 eq) was stirred for 4 h at r.t. under argon. The solvent was evaporated to give 8 as a white solid in quantitative yield [7]. LC: tr 3.67’ (purity of >95%), MS: 448 [M+H]+.
4-trans-Pentyl-cyclohexanecarboxylic acid [1-(4-hydroxy-benzyl)-piperidin-4-yl]-(4-pyridin-4-yl-benzyl)-amide (10)
To a solution of compound 8 (63 mg, 0.131 mmol, 1 eq) in CH2Cl2 (10 mL) was added 4-hydroxy-benzaldehyde (64 mg, 0.524 mmol, 4 eq) and NaBH(OAc)3 (138 mg) and stirring was continued at r.t. under argon for 12 h. The reaction mixture was washed once with saturated NaHCO3 solution, dried over MgSO4 followed by chromatographic purification to give 42.3 mg (58%) of the final compound 10 as a white solid. LC: tr 4.33’; MS: 555 [M+H]+; 1H-NMR: 8.68-8.47 (m, 2H), 7.66-6.61 (m, 10H), 5.27 (s, 1H), 4.59, 4.56 (s, 2H, benzylic methylene of both rotamers) 3.86 (m, 1H), 3.19 (s, 2H), 3.07-2.79 (m, 2H), 2.62-2.41 (m, 1H), 2.31-0.59 (m, 26H).
Acknowledgements
The authors would like to thank Dr. T. Weller and Dr. F. Spindler for helpful discussions, Prof H. Ramuz for his interest in the present work and S. Meyer for performing the plasmepsin II screening.
References
- (a) Bell, A. I. Drugs 2000, 3, 310–317. (b) Bell, A. Curr. Opin. Anti-infect. Invest. Drugs 2000, 2, 63–70.
- Rosenthal, P. J.; Sijwali, P.S.; Singh, A.; Shenai, B. R. Curr. Pharm. Des. 2002, 8, 99–110.
- http://sites.huji.ac.il/malaria/hemoglobindigest.html
- Rosenthal, P. J. Antimalarial Chemotherapy: Mechanism of Action, Resistance, and New Directions in Drug Discovery; Humana Press: New Jersey, 2001. [Google Scholar]
- Berry, C. Curr. Opin. Drug Disc. Dev. 2000, 3, 624–629.
- Boss, C.; Richard-Bildstein, S.; Weller, T.; Fischli, W.; Meyer, S.; Binkert, C. Curr. Med. Chem. 2003, 10, 883–907.
- Actelion Pharmaceuticals Ltd (Boss, C.; Fischli, W.; Meyer, S.; Richard-Bildstein, S.; Weller, T.), 2002, WO 02/24649
- (a) Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457–2483. (b) Watanabe, T.; Miyaura, N.; Suzuki, A. Synlett 1992, 207–210. (c) Gong, Y.; Pauls, H.W. Synlett 2000, 829–831.
- Abdel-Magid, A. F.; Carson, K. G.; Harris, B. D.; Maryanoff, C. A.; Shah, R. D. J. Org. Chem. 1996, 61, 3849–3862.
- Matayoshi, E. D.; Wang, G. T.; Krafft, G.A.; Erickson, J. Science 1990, 247, 954–958.
- Mueller, R. Diploma Thesis, University of Applied Sciences Basel, Muttenz, 2002.
- Taber, D. F.; Deker, P. B.; Gaul, M. D. J. Am. Chem. Soc. 1987, 109, 7488–7494.
- Sample Availability: Not available.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Mueller, R.; Huerzeler, M.; Boss, C. Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents. Molecules 2003, 8, 556-564. https://doi.org/10.3390/80700556
AMA Style
Mueller R, Huerzeler M, Boss C. Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents. Molecules. 2003; 8(7):556-564. https://doi.org/10.3390/80700556
Chicago/Turabian StyleMueller, Reto, Marianne Huerzeler, and Christoph Boss. 2003. "Synthesis of Plasmepsin II Inhibitors – Potential Antimalarial Agents" Molecules 8, no. 7: 556-564. https://doi.org/10.3390/80700556