Phthalimides: Supramolecular Interactions in Crystals, Hypersensitive Solution 1H-NMR Dynamics and Energy Transfer to Europium(III) and Terbium(III) States

Abstract
:Introduction



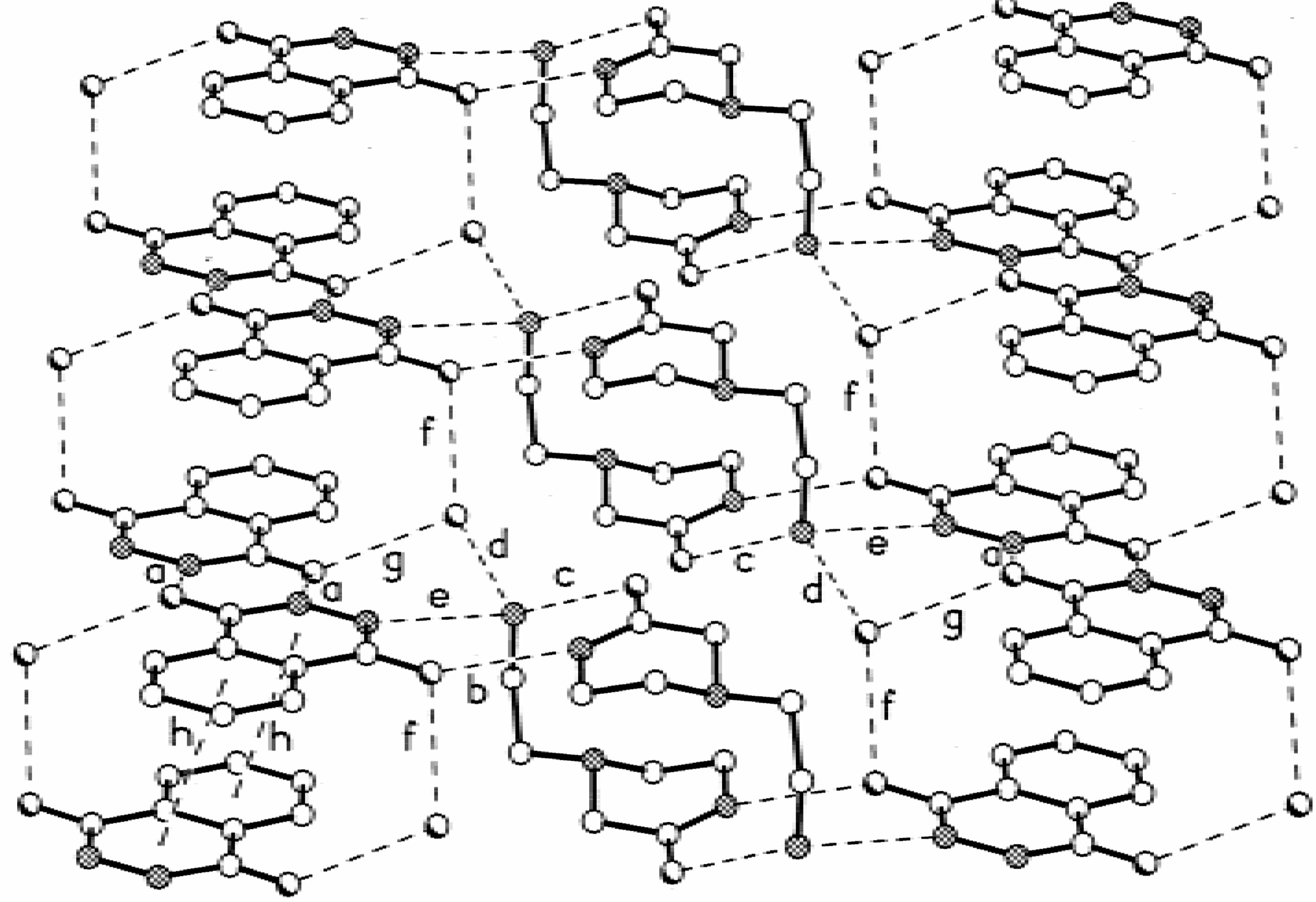
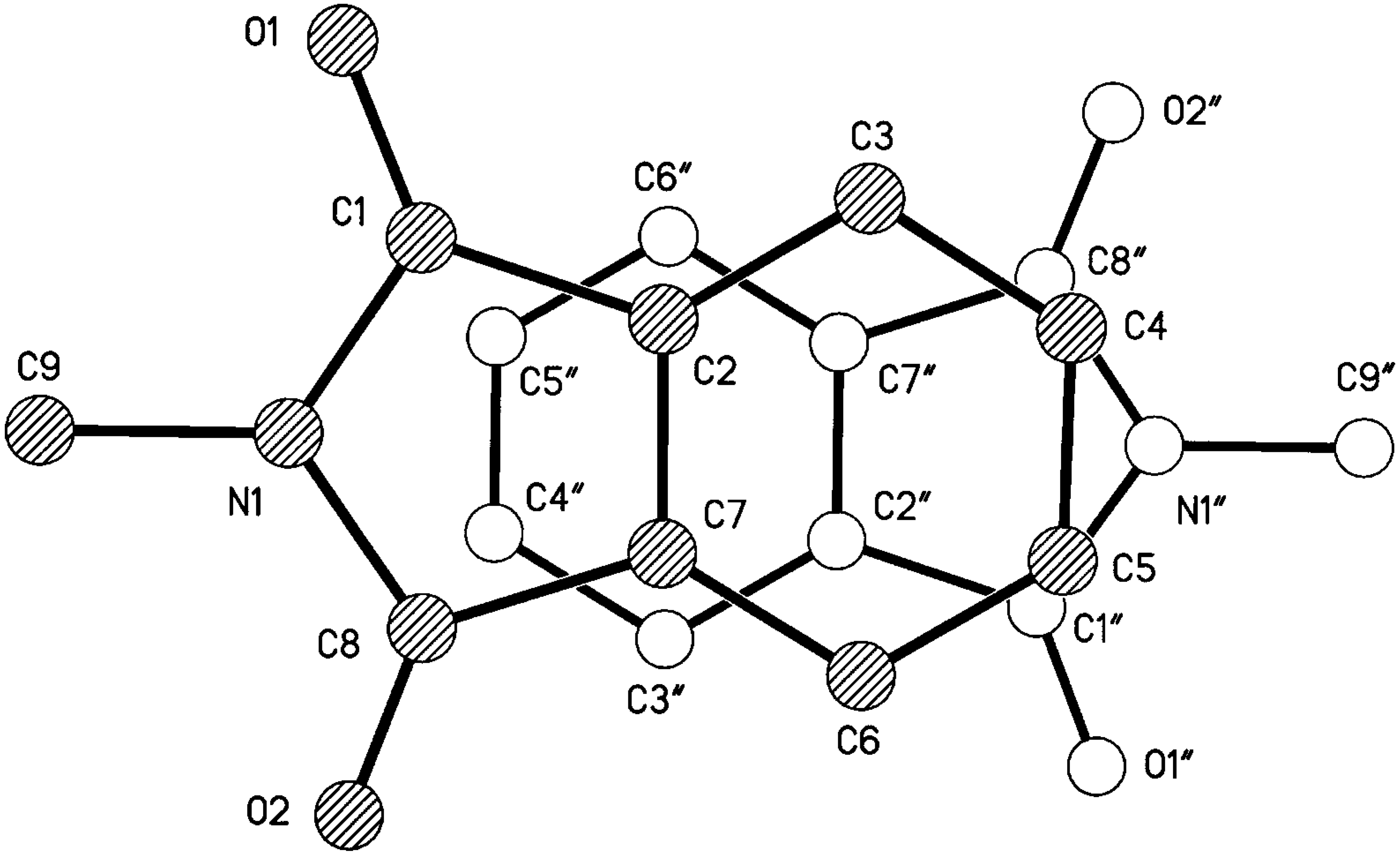
Results and Discussion
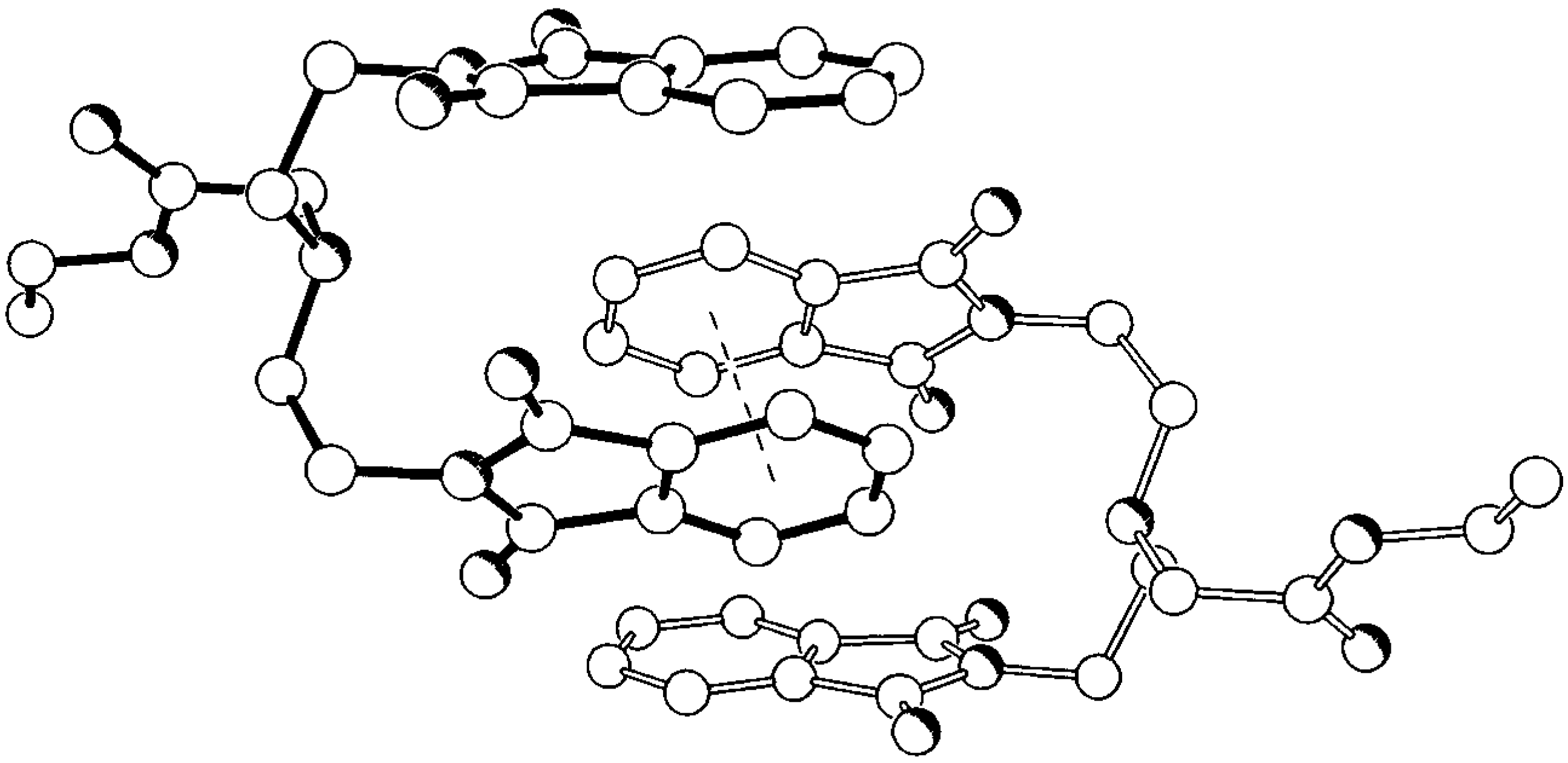
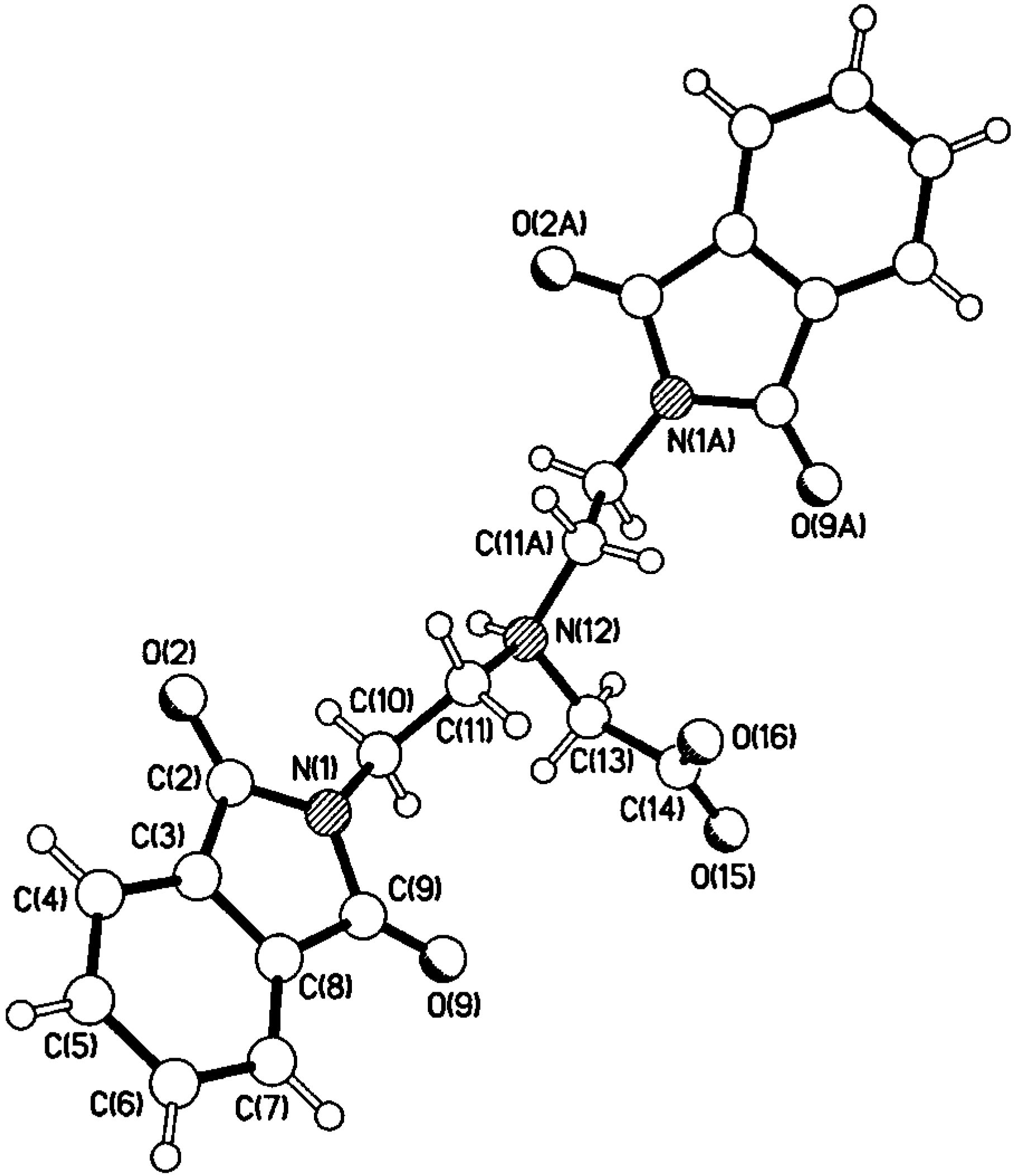
Compounds 5 and 6




Reaction of 7 with Hydrazine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
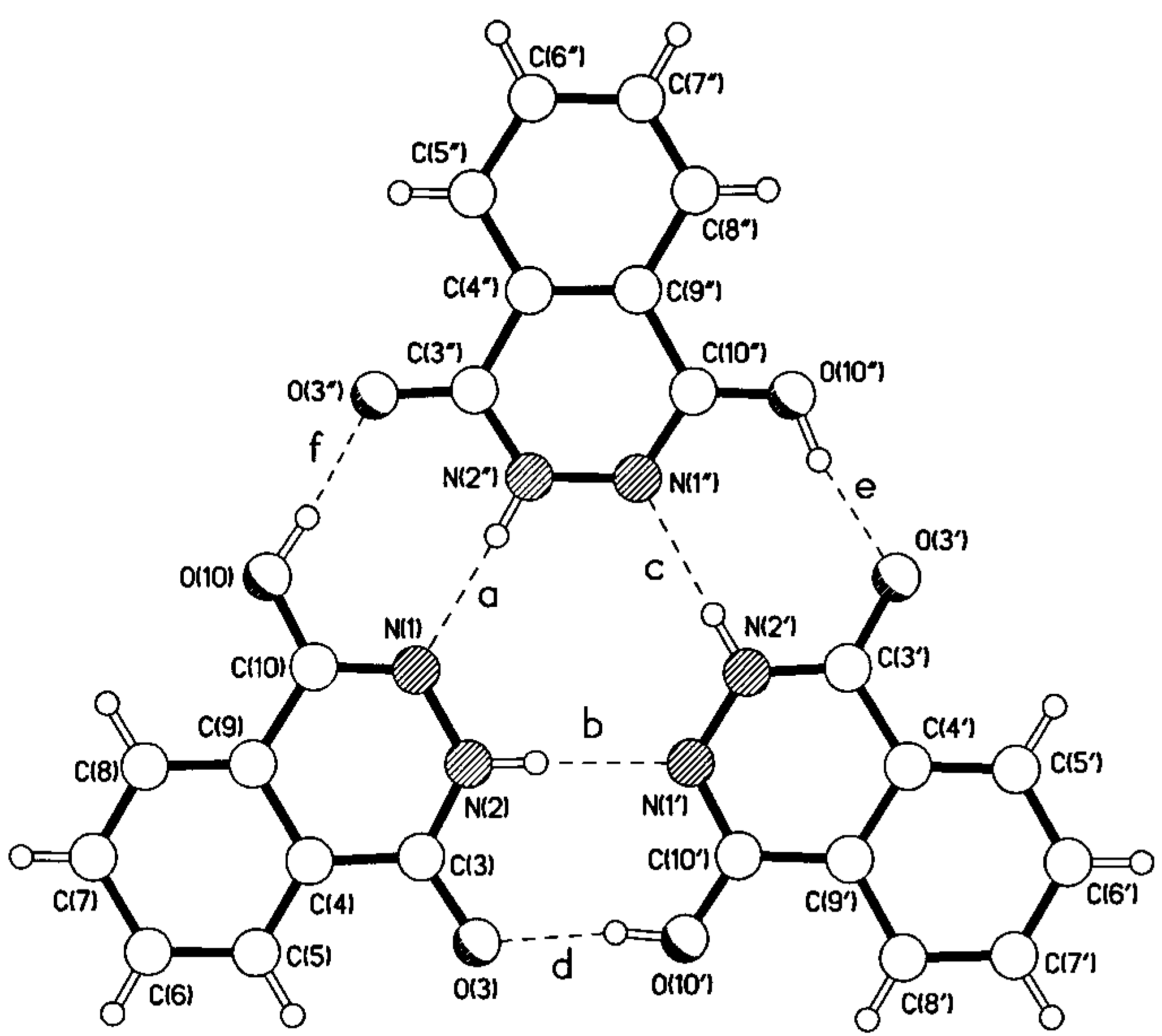
| a | 2.85 | 1.96 | 173 |
| b | 2.84 | 1.94 | 177 |
| c | 2.84 | 1.95 | 170 |
| d | 2.68 | 1.79 | 171 |
| e | 2.68 | 1.78 | 167 |


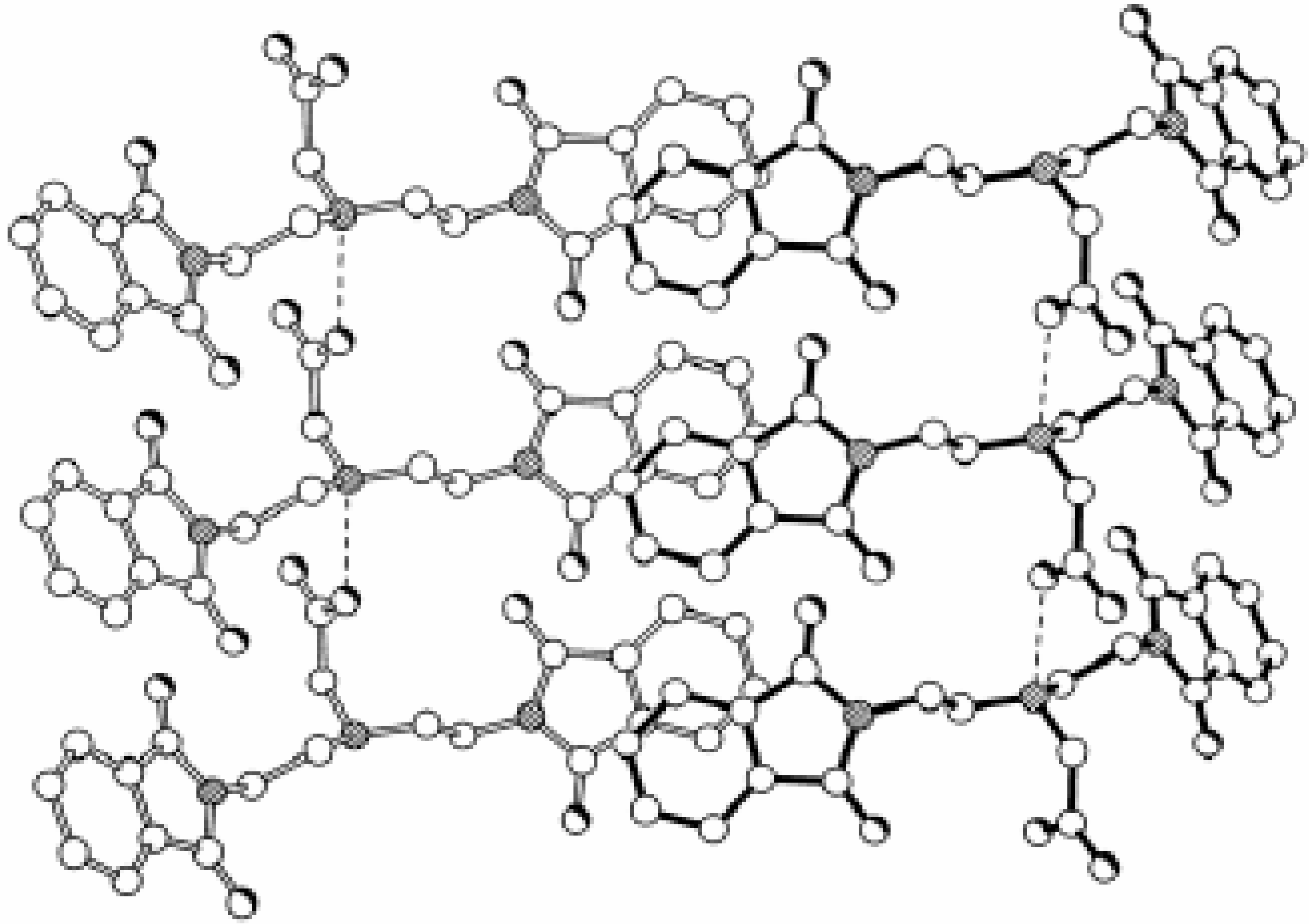
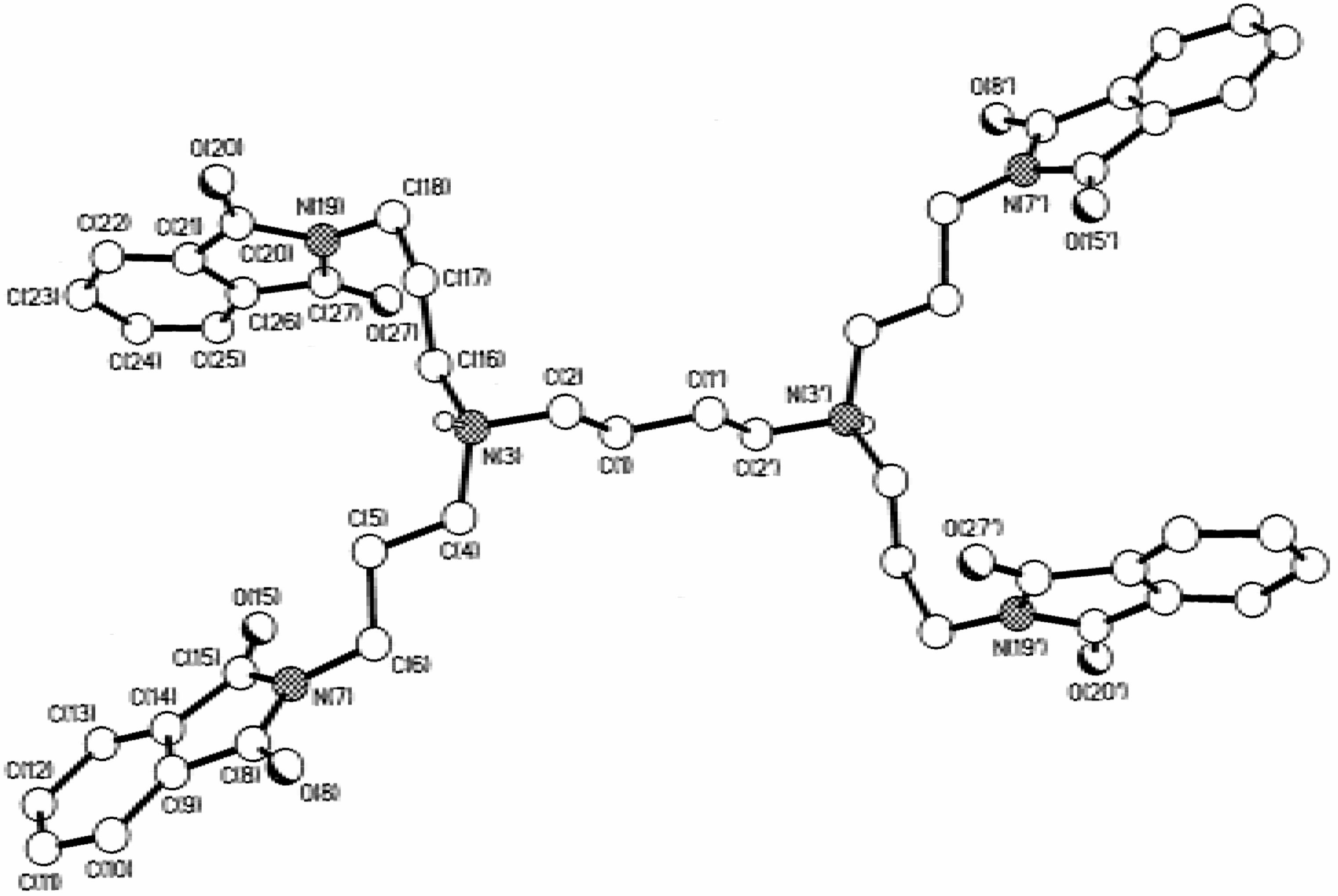
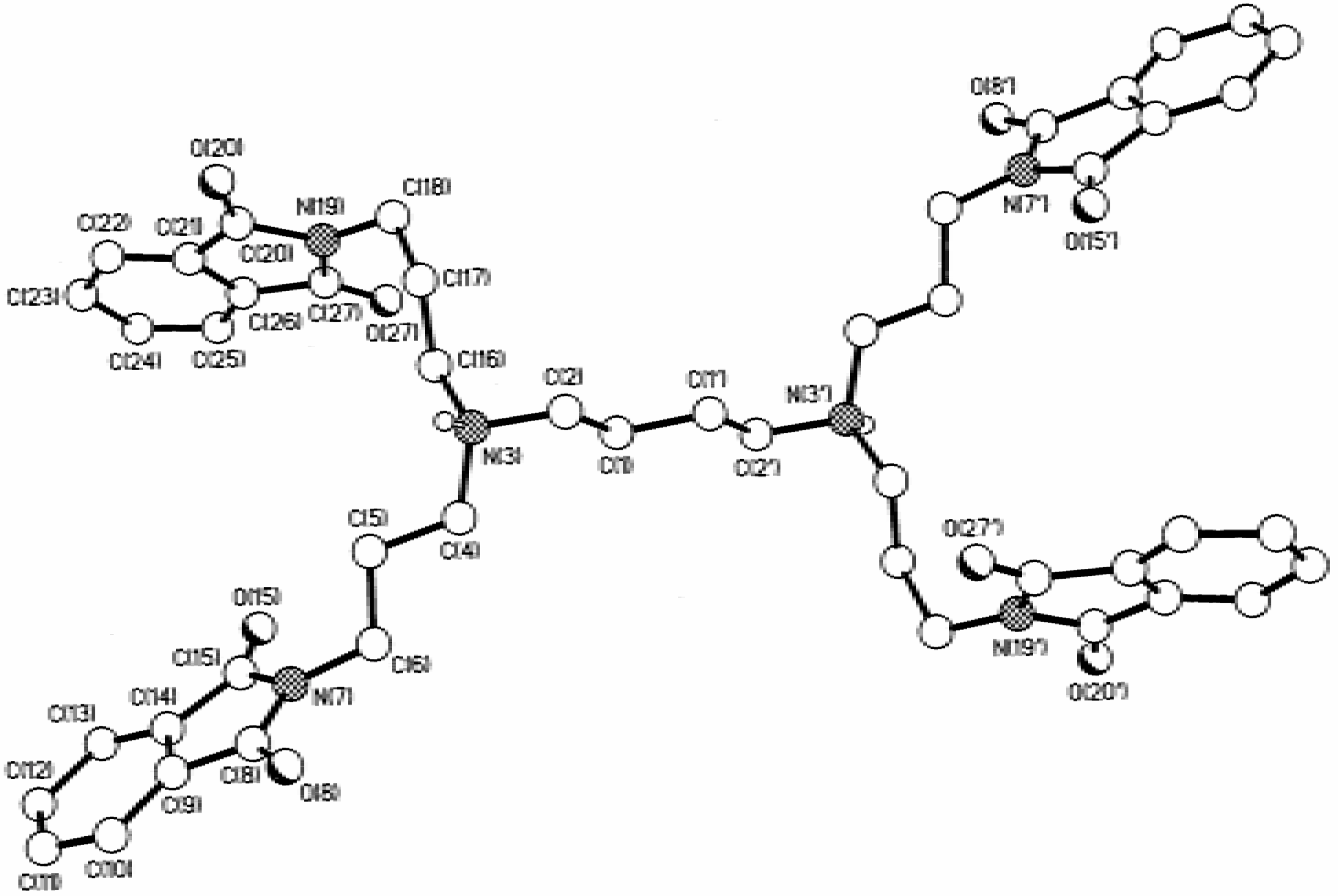
Dendrimers


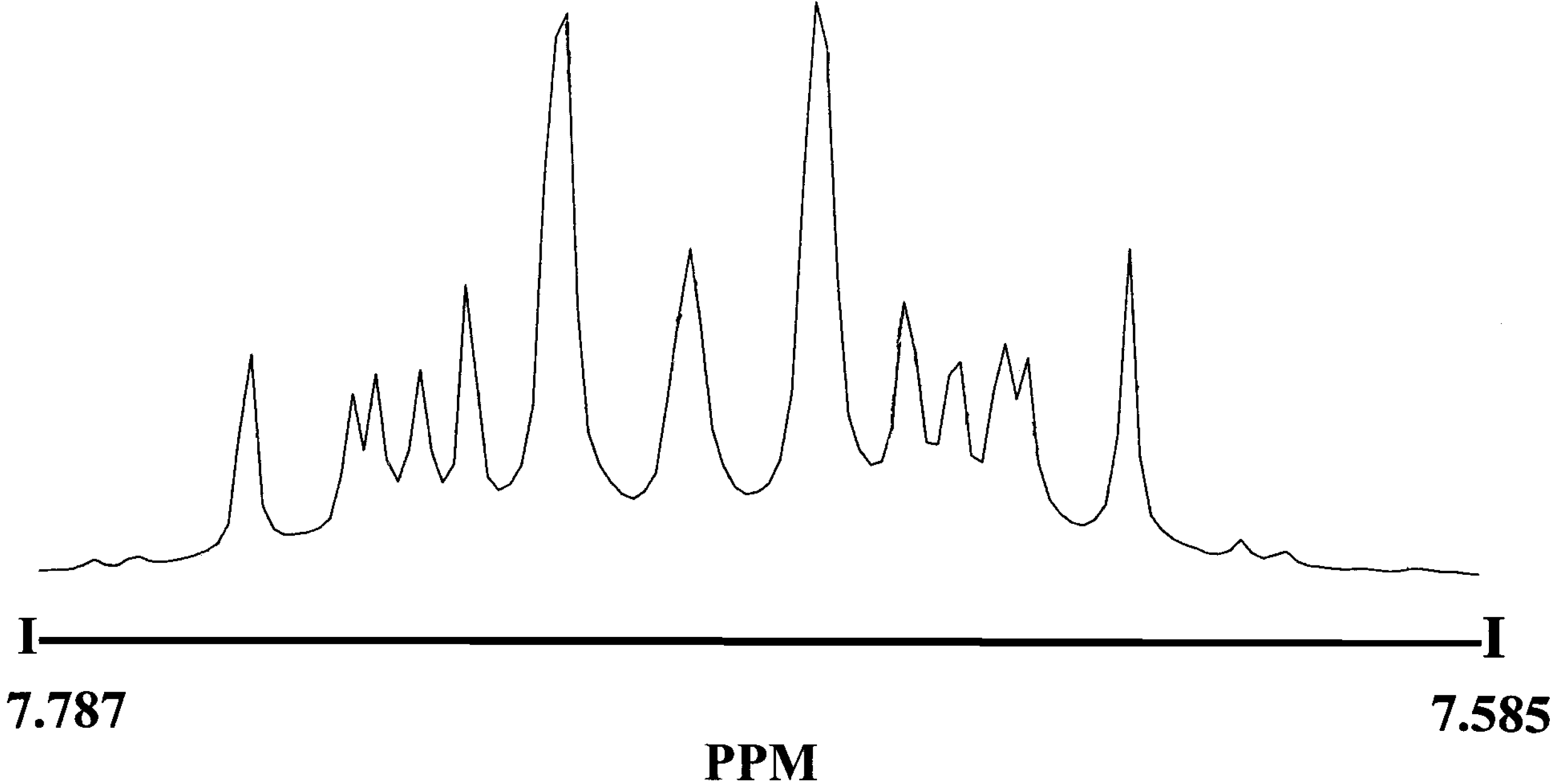
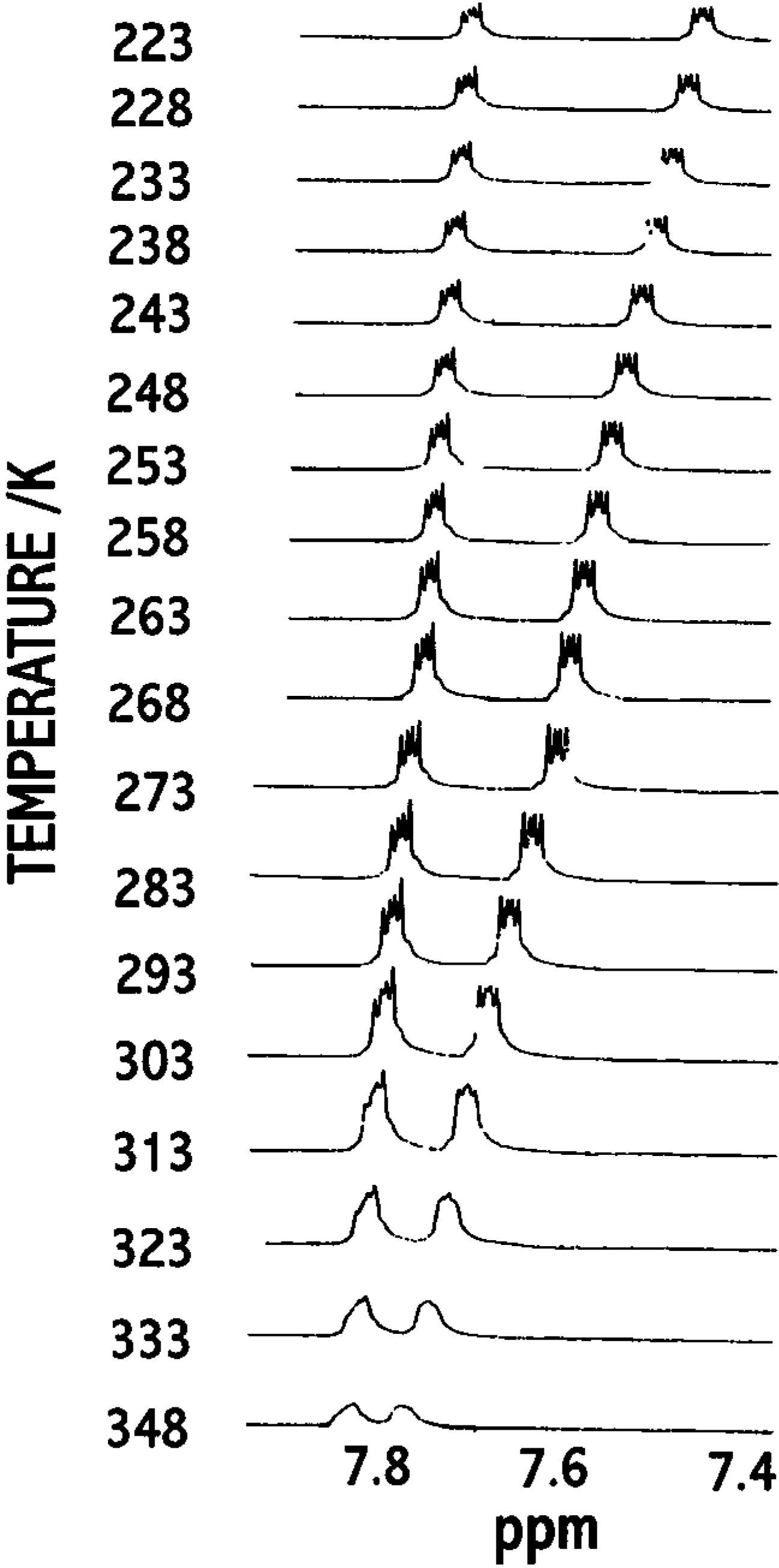
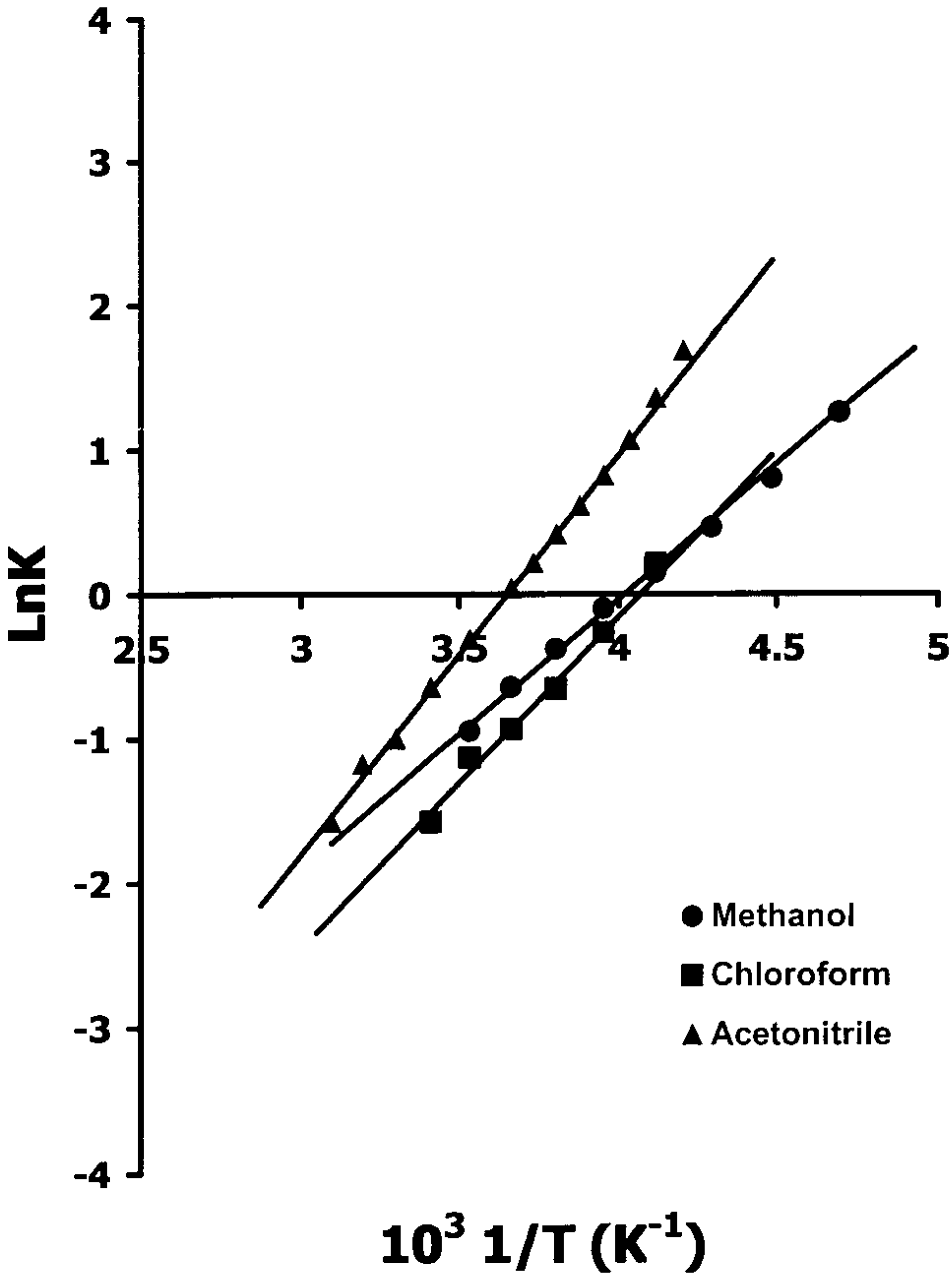
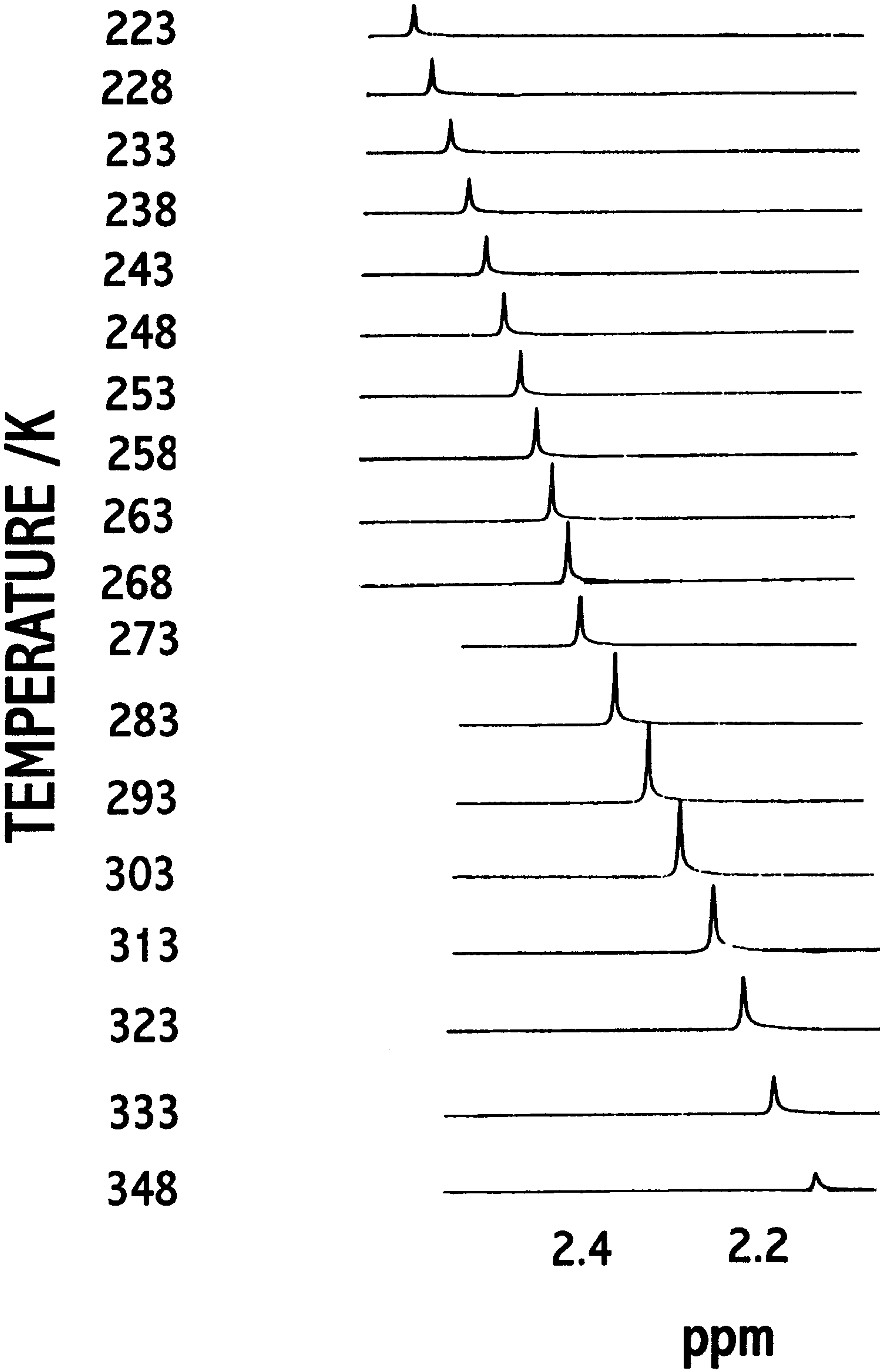
Hypersensitive 1H -NMR of 3 and 5

- δu = upper chemical shift limit;
- δl = lower chemical shift limit;
- δ = temperature dependent chemical shift of hypersensitive reference peak (the most intense one);
- ΔH# = Process enthalpy;
- ΔS# = Process entropy


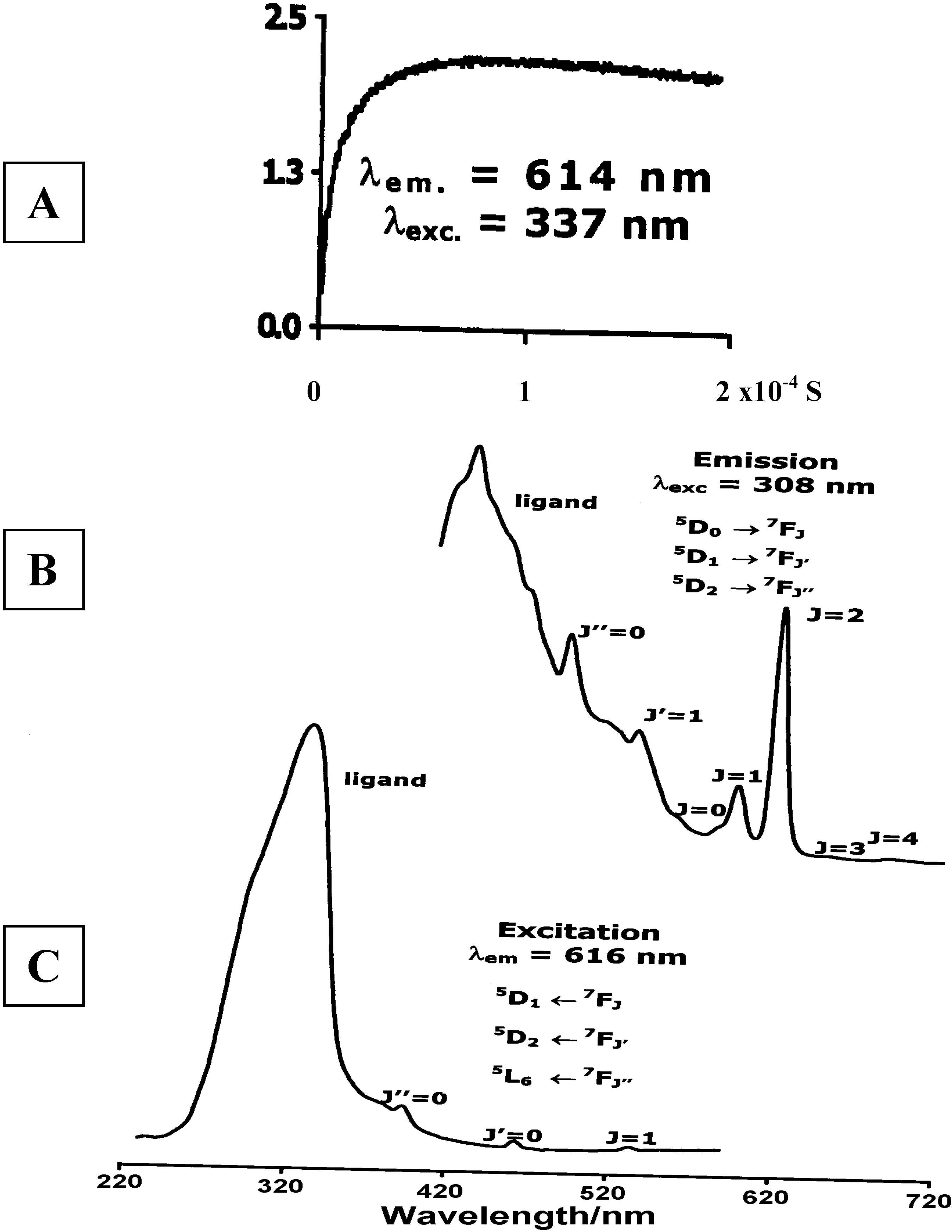
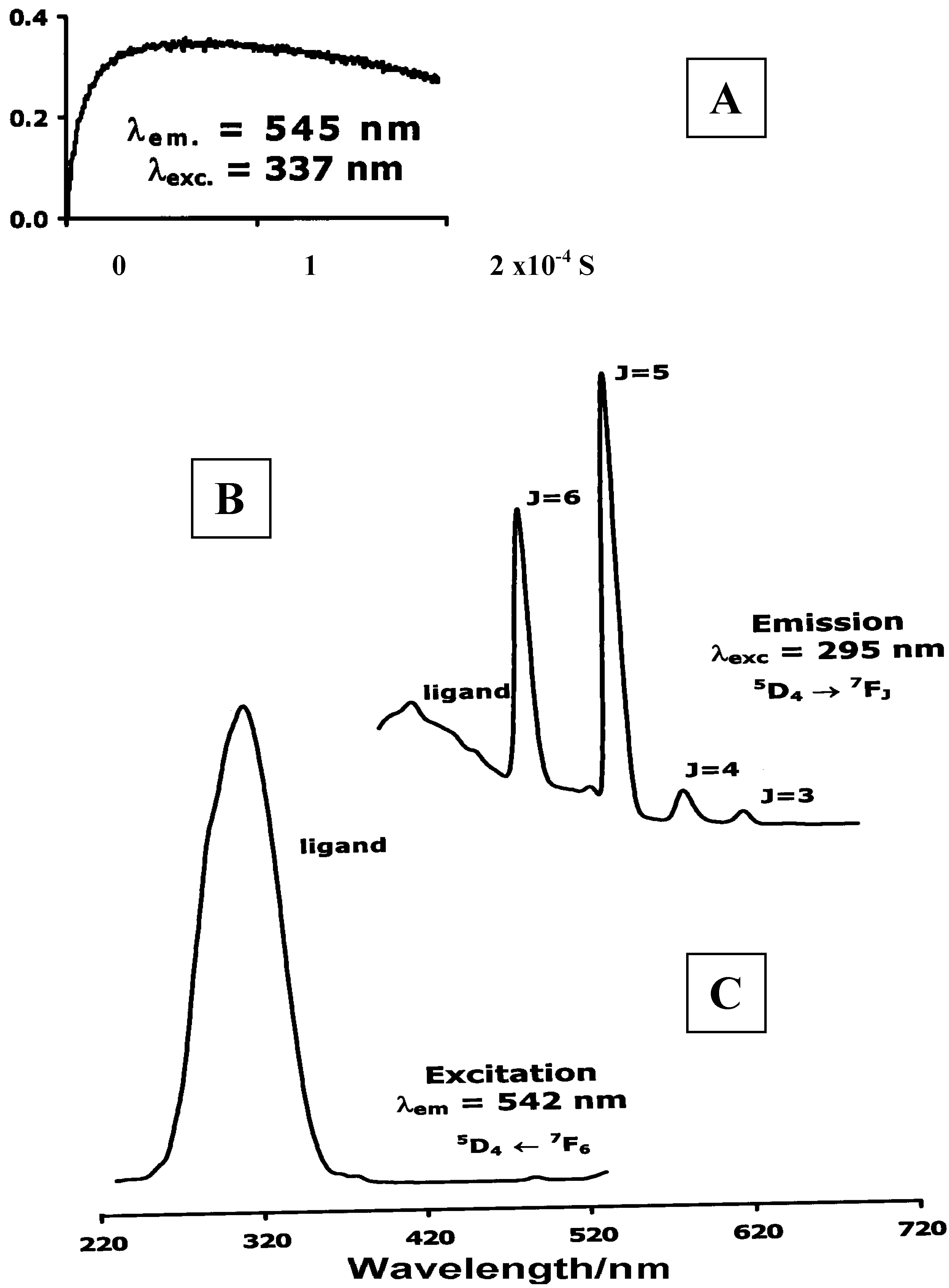
Luminescence of Europium(III) and Terbium(III) complexes of Dendrimer 10
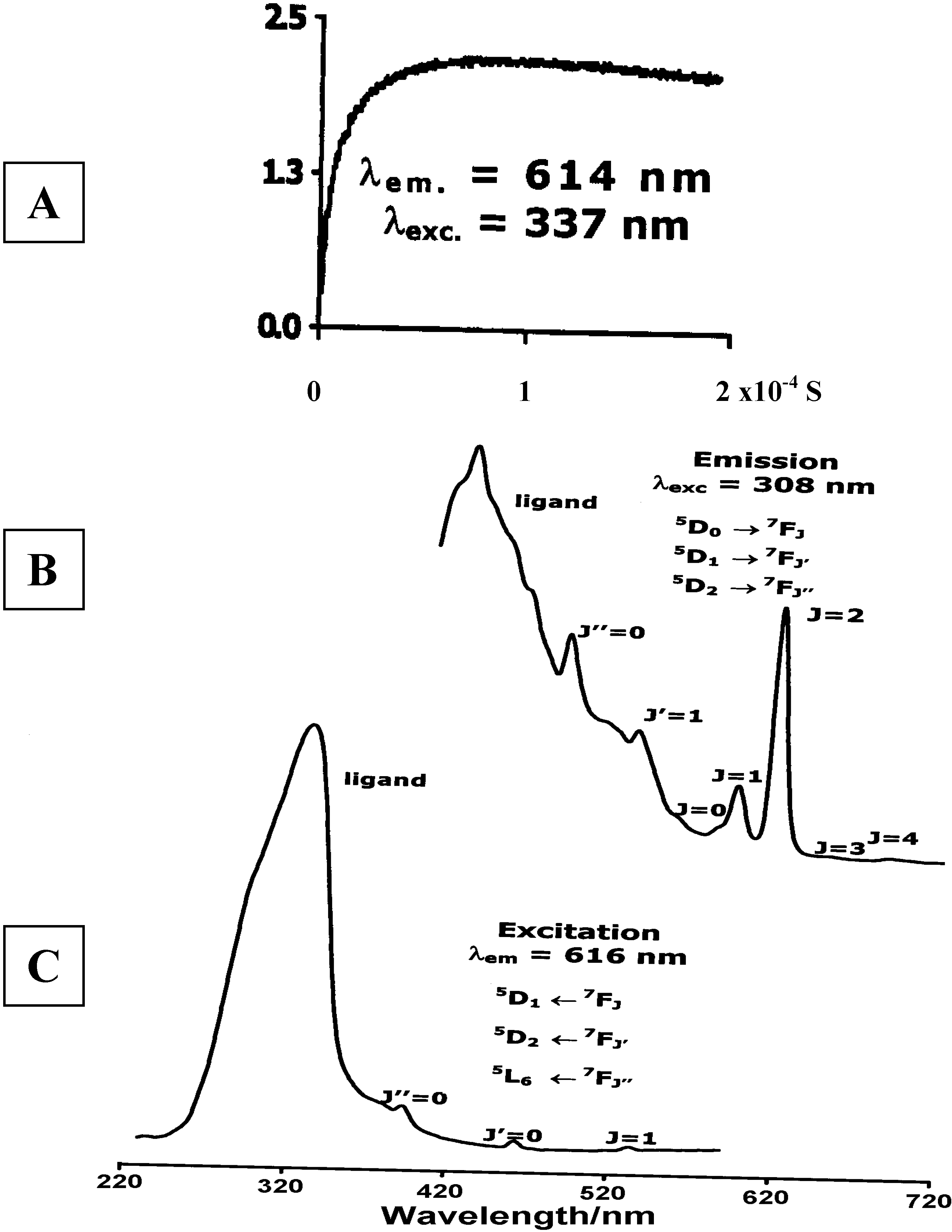

Concluding Remarks
Experimental
General
Crystallography
| Compound | 3 | 5 | 6.H2O | 8.H2O | 9 | [H210][ClO4]2· CH3CN |
|---|---|---|---|---|---|---|
| Formula | C42H36N6O8 | C24H23N3O6 | C22H19N3O6.H2O | C6H14N3O· C8H5N2O2·H2O | C8H6N2O2 | [C48H50N6O8]· [ClO4]2.CH3CN |
| Formula Weight | 752.77 | 449.5 | 439.4 | 323.4 | 162.2 | 1078.89 |
| Lattice type | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Orthorhombic |
| Space group | ||||||
| Symbol, No. | Pī, 2 | Pī, 2 | P21/m, 11 | P21/c, 14 | Cc, 9 | Pnna, 52 |
| Cell | ||||||
| dimensions | ||||||
| a/Å | 9.773(1) | 9.134(2) | 5.429(1) | 15.118(2) | 22.472(2) | 24.886(5) |
| b/Å | 11.755(1) | 11.131(2) | 22.306(5) | 7.590(2) | 13.931(2) | 25.703(4) |
| c/ Å | 9.0341(8) | 12.012(2) | 9.276(2) | 14.969(3) | 7.093(2) | 7.825(1) |
| α/deg | 111.740(9) | 64.48(2) | - | - | - | 90 |
| β/deg | 94.236(9) | 87.98(2) | 93.35(2) | 1123.89 | 101.49(1) | 90 |
| γ/deg | 81.31(1) | 85.52(2) | - | - | - | 90 |
| V/ Å3 | 952.8(2) | 1098.7(4) | 1121.4(5) | 1570.4(5) | 2176.0(6) | 5005(2) |
| Z | 1 | 2 | 2[a] | 4 | 12[b] | 4 |
| F(000) | 394 | 472 | 460 | 688 | 1008 | 2256 |
| Radiation | Mo-Kα | Cu-Kα | Mo-Kα | Cu-Kα | Mo-Kα | Cu-Kα |
| μ/mm-1 | 0.093 | 0.82 | 0.10 | 0.85 | 0.11 | 1.845 |
| R1 | 0.043 | 0.049 | 0.084 | 0.05 | 0.041 | 0.0886 |
| wR2 | 0.11 | 0.134 | 0.189 | 0.134 | 0.103 | 0.2283 |
Preparation of dendrimer N,N,N',N'-tetrakis(2-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-ethyl)-1,2-ethyl-diamine (3).
{Bis-[2-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-ethy]-amino}-acetic acid ethyl ester (5)
{Bis-[2-(1,3-dioxo-1,3-dihydro-isoindol-2-yl)-ethyl]-aminium} acetate (6)
{(4-(2-Aminium-ethyl)-piperazin-2-one)(2-hydro-phthalazinate-1,4-dione)} (8)
4-Hydroxy-2H-phthalazin-1-one (9)
Dendrimers N,N,N',N'-tetrakis(3-(1,3-dioxo-1,3-dihydroisoindol-2-yl)-propyl)-1,4-butanediamine (10), [4,17-bis(3-(1,3-dioxo-1,3-dihydroisoindol-2-yl)-propyl)-8,13-bis[3-[bis(3-aminopropyl)-amino]-propyl]-4,8,13,17-tetraazaeicosane-1,20-diamine] (11) and 1,4-diaminobutane[16]:(1,3-dioxo-1,3-dihydroisoindol-2-yl)-propylamine (12)
Compound [H210][ClO4)2·0.5H2O
Lanthanide complexes of 10
Compounds Ln5(H11)(NO3)16·solvent
Supplementary Material

| a (N1): | 2.81, | 1.95, | 159. |
| b (N11): | 2.75, | 1.94, | 147. |
| c (N19): | 2.90, | 2.04, | 160. |
| d (N19): | 2.76, | 1.95, | 148. |
| e (N19): | 2.90, | 2.05, | 157. |
| f (O20): | 2.70, | 1.80, | 172. |





Acknowledgements
References and Notes
- Barrett, D. M. Y.; Kahwa, I. A.; Mague, J. T.; McPherson, G. L. J. Org. Chem. 1995, 60, 5946.
- Barrett, D. M. Y.; Kahwa, I. A.; Williams, D. J. Acta Crystallogr. Sect. C. 1996, 52, 2069.
- Barrett, D. M. Y.; Kahwa, I. A.; Radüchel, B.; White, A. J. P.; Williams, D. J. J. Chem. Soc. Perkin Trans. 2 1998, 1851.
- Silverstein, R. M.; Webster, F. X. Spectrometric Identification of Organic Compounds, Sixth edition; John Wiley & Sons, Inc: New York, 1997; p. 174. [Google Scholar]
- Gawronski, J.; Kazmierczak, F.; Grownska, K.; Rychlewska, U.; Nordé, B.; Holmén, A. J. Am. Chem. Soc. 1998, 120, 12083.
- Hamilton, D. G.; Lynch, D. E.; Byriel, K. A.; Kennard, C. H. L. Austr. J. Chem. 1997, 50, 439.
- Hamilton, D. G.; Feeder, N.; Prodi, L.; Teat, S. J.; Clegg, W.; Sanders, J. K. M. J. Am. Chem. Soc. 1998, 120, 1096.
- Schirling, D.; Decep, J. B.; Baltzer, S.; Pirion, F.; Wagner, J.; Hornperger, J. M.; Heydt, J. C.; Jung, M. J.; Danzin, C.; Weiss, R.; Fisher, J.; Mitshler, A.; De Cian, A. J. Chem. Soc. Perkin Trans. 1 1992, 1053.
- Jun-Li, T.; Ruan-Hua, D.; Yuen-Ping, L.; Zhong-Yuan, Z.; Li, L.; Kai-Bei, Y. Chinese J. Struct. Chem. (Jegon Huaxue) 1991, 10, 187.
- Gajadhar-Plummer, A. S. Ph.D. Thesis, University of the West Indies, Mona, 1999.
- (a) Barrett, D. M. Y. Ph.D. Thesis, University of the West Indies, Mona, 1997. (b) Howell, R. C. Ph.D. Thesis, University of the West Indies, Mona, 1997.
- (a) Howell, R. C.; Kahwa, I. A.; White, A. J. P.; Williams, D. J. J. Chem. Soc. Dalton Trans. 1996, 961. (b) Howell, R. C.; Spence, K. V. N.; Kahwa, I. A.; Williams, D. J. J. Chem. Soc. Dalton Trans. 1998, 2727.
- Williams, L. A. D.; Howell, R. C.; Young, R.; Kahwa, I. A. Comparative Biochem. Physiol. Part C 2001, 128, 119.
- (a) Latva, M.; Mäkinen, P.; Kulmala, S.; Haapakka, K. J. Chem. Soc. Faraday Trans. 1996, 92, 3321. (b) Chen, J.; Selvin, P. R. J. Am. Chem. Soc. 2000, 122, 657. (c) Fatin-Rouge, N.; Tóth, É.; Perret, D.; Backer, R. H.; Merbach, A. E.; Bünzli, J.-C. G. J. Am. Chem. Soc. 2000, 122, 10810.
- Bruce, J. I.; Parker, D.; Tozer, D. Chem. Commun. 2000, 2250.
- (a) Bornhop, D. J.; Hubbard, D. S.; Houlne, M. P.; Adair, C.; Kiefer, G. E.; Pence, B.C.; Morgan, D. L. Anal. Chem. 1999, 71, 2607. (b) Horrocks Jr, W. deW.; Bolender, J. P.; Smith, W.D.; Supkowski, R. M. J. Am. Chem. Soc. 1997, 119, 5972.
- (a) Aime, S.; Botta, M.; Fasano, M.; Terreno, E. Chem. Soc. Rev. 1998, 27, 19. (b) Cohen, S. M.; Xu, J.; Radkov, E.; Raymond, K. N.; Botta, M.; Barge, A.; Aime, S. Inorg. Chem. 2000, 39, 5747. (c) Ellison, J. J.; McMurry, T. J.; Lauffer, R. B. Chem. Rev. 1999, 99, 2293.
- (a) Zitha-Boven, E.; Elst, L. V.; Muller, R. N.; van Bekkum, H.; Peters, J. A. Eur. J. Chem. 2001, 3101. (b) Merbach, A. E.; Tóth, É (Eds.) The Chemistry of Contrast Agents in Magnetic Resonance Imaging; John Wiley: Chichester, U.K, 2001.
- (a) Doyle, J. D.; Harriman, A.; Newport, G. L. J. Chem. Soc. Perkin Trans. 1979, 2, 799. (b) Zenner, B.; Bender, M. L. J. Am. Chem. Soc. 1961, 83, 2267.
- (a) Epsetein, D. M.; Chappell, L. L.; Khalili, H.; Supkowski, R. M.; Horrocks Jr., W. deW.; Morrow, J. R. Inorg. Chem. 2000, 39, 2130. (b) Gomez-Tangle, P.; Yatsimirsky, A. K. J. Chem. Soc. Dalton Trans. 2001, 2663. (c) Bligh, S. W. A.; Choi, N.; Evagorou, E. G.; McPartlin, M.; White, K. N. J. Chem. Soc. Dalton Trans. 2001, 3169.
- Thompson, M. K.; Vuchkov, M.; Kahwa, I. A. Inorg. Chem. 2001, 40, 4332.
- Schneider, H.-J. Angew. Chem. Int. Ed. Engl. 1991, 30, 1417.
- (a) Ng, C. Y.; Motekaitis, R. J.; Martel, A. E. Inorg. Chem. 1979, 18, 2982. (b) Buckingham, D. A.; Edwards, J. D.; McLaughlin, G. M. Inorg. Chem. 1982, 21, 2770.
- Kozlecki, T.; Samyn, C.R.; Alder, W.; Green, P. G. J. Chem. Soc. Perkin Trans 2 2001, 243.
- Padies, H. H. Bunsenges. Phys. Chem. 1992, 96, 1027.
- Becker, D.; Botoshansky, M.; Gasper, N.; Herbstein, F. H.; Karni, M. Acta Crystallogr. Sec. 1998, B54, 671.
- Gane, P. A.; Boles, M. O. Acta Crystallogr. 1979, B35, 2664.
- Isotopic abundance patterns were calculated with Isopro V 2.1 written by Yergey, J. A. Sheffield University (UK) and available at: http://www.chem.schef.ac.uk./webelements.cgi$isot.
- Feeney, J.; Walker, S. M. J. Chem. Soc. (A) 1966, 1148.
- Platas-Iglesias, C.; Piguet, C.; André, N.; Bünzli, J.-C. G. J. Chem. Soc. Dalton Trans. 2001, 3084.
- Rigault, S.; Piguet, C. J. Am. Chem. Soc. 2001, 122, 9304.
- (a) Dexter, L. J. Chem. Phys. 1953, 21, 836. (b) Förster, Th. Ann. Phys. (Leipzig) 1948, 2, 55.
- (a) Newkome, G. R.; Moorefield, C. N.; Vögtle, F. Dendritic Molecules Concepts, Synthesis, Perspectives; VCH: Weinheim, Germany, 1996. [Google Scholar] (b) Newkome, G. R.; He, E.; Moorefield, C. W. Chem. Rev. 1999, 99, 1689.
- (a) Grayson, S. M.; Fréchet, J. M. Chem. Rev. 2001, 101, 3819. (b) Zeng, F.; Zimmerman, S.C. Chem. Rev. 1997, 97, 1681. (c) Astruc, F.; Chardac, F. Chem. Rev. 2001, 101, 2991. (d) Bosman, A. W.; Janssen, H. M.; Meijer, E. W. Chem. Rev. 1999, 99, 1665. (e) Schenning, J.; Meijer, E. W.; Bässler, H. J. Phys. Chem. A. 2001, 105, 10220. (f) Meskers, S. C. J.; Bender, M.; Hübner, J.; Romanovskii, Yu. V.; Oestreich, M.; Weener, A. P. H. J.-W.; van Dongen, J. L. J.; Meijer, E. W. J. Am. Chem. Soc. 1999, 121, 10346.
- Tavakovic, I.; Miller, L. L.; Duan, R. G.; Tully, D. C.; Tomalia, D. C. Chem. Mater. 1997, 9, 736.
- Sample Availability: Samples of compounds 3, 5, 10 and 11 are available from the authors on request.
© 2003 by MDPI ( http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Howell, R.C.; Edwards, S.H.; Gajadhar-Plummer, A.S.; Kahwa, I.A.; McPherson, G.L.; Mague, J.T.; White, A.J.P.; Williams, D.J. Phthalimides: Supramolecular Interactions in Crystals, Hypersensitive Solution 1H-NMR Dynamics and Energy Transfer to Europium(III) and Terbium(III) States. Molecules 2003, 8, 565-592. https://doi.org/10.3390/molecules8070565
Howell RC, Edwards SH, Gajadhar-Plummer AS, Kahwa IA, McPherson GL, Mague JT, White AJP, Williams DJ. Phthalimides: Supramolecular Interactions in Crystals, Hypersensitive Solution 1H-NMR Dynamics and Energy Transfer to Europium(III) and Terbium(III) States. Molecules. 2003; 8(7):565-592. https://doi.org/10.3390/molecules8070565
Chicago/Turabian StyleHowell, Robertha C., Selvin H. Edwards, Alison S. Gajadhar-Plummer, Ishenkumba A. Kahwa, Gary L. McPherson, Joel T. Mague, Andrew J. P. White, and David J. Williams. 2003. "Phthalimides: Supramolecular Interactions in Crystals, Hypersensitive Solution 1H-NMR Dynamics and Energy Transfer to Europium(III) and Terbium(III) States" Molecules 8, no. 7: 565-592. https://doi.org/10.3390/molecules8070565