Atom, Atom-Type, and Total Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to QSPR/QSAR Studies of Organic Compounds

Abstract
:Introduction
Theoretical Approach
Local (Atom) Linear Indices of the “Molecular Pseudograph’s Atom (Vertex) Adjacency Matrix”

= Lii if i = j
= 0 otherwise

{kind=link}
{kind=link}
{kind=link}
{kind=link}
 2-Amino-benzaldehyde MolecularStructure |  Molecular Pseudograph (G) (hydrogen suppressed-pseudograph) | X = [O1, C2, C3, C4, C5, C6, C7, C8, N9] Molecular Vector: X∈ℜ9 In the definition of the X, as molecular vector, the chemical symbol of the element is used to indicate the corresponding electronegativity value. That is: if we write O it means χ(O), oxygen Mulliken electronegativity or some atomic property, which characterizes each atom in the molecule. Therefore, if we use the canonical basis of 9, the coordinates of any vector X coincide with the components of that molecular vector [X] = [3.17, 2.63, 2.63, 2.63, 2.63, 2.63, 2.63, 2.63, 2.33] [X]: column vector of coordinates of X in the Canonical base of R9 (a nx1 matrix) | ||||||
 |  | |||||||
| Atom linear indices of first order is a linear map; f1(xi): ℜn→ ℜn such that, f1(O1, C2, C3, C4, C5, C6, C7, C8, N9) = (2C2, 2O1+1C3, 1C2 +1C3+1C4+1C8, 1C3+1C4+1C5, 1C4+1C5+1C6, 1C5+1C6+1C7, 1C6+1C7+1C8, 1C3+1C7+1C8+1N9, 1C8) = (5.26, 8.97, 10.52, 7.89, 7.89, 7.89, 7.89, 10.22, 2.63) and whole-molecule linear indices of first order is a linear functional;  =f1(O1) + f1(C2) + f1(C3) + f1(C4) + f1(C5) + f1(C6) + f1(C7) + f1(C8) + f1(N9)= 69.16 =f1(O1) + f1(C2) + f1(C3) + f1(C4) + f1(C5) + f1(C6) + f1(C7) + f1(C8) + f1(N9)= 69.16 | ||||||||
| Local and total linear indices of order 0-5 (k = 0-5) | ||||||||
| Atom (i) | f0(xi) | f1(xi) | f2(xi) | f3(xi) | f4(xi) | f5(xi) | ||
| O1 | 3.17 | 5.26 | 17.94 | 42.08 | 146.96 | 400.72 | ||
| C2 | 2.63 | 8.97 | 21.04 | 73.48 | 200.36 | 676.25 | ||
| C3 | 2.63 | 10.52 | 37.6 | 116.2 | 382.33 | 1193.57 | ||
| C4 | 2.63 | 7.89 | 26.3 | 87.57 | 277.41 | 894.29 | ||
| C5 | 2.63 | 7.89 | 23.67 | 73.64 | 234.55 | 739.87 | ||
| C6 | 2.63 | 7.89 | 23.67 | 73.34 | 227.91 | 721.81 | ||
| C7 | 2.63 | 7.89 | 26 | 80.93 | 259.35 | 820.73 | ||
| C8 | 2.63 | 10.22 | 31.26 | 105.08 | 333.47 | 1080.23 | ||
| N9 | 2.33 | 2.63 | 10.22 | 31.26 | 105.08 | 333.47 | ||
| Total | 23.91 | 69.16 | 217.7 | 683.58 | 2167.42 | 6860.94 | ||
Total (Whole-Molecule) Linear Indices of the “Molecular Pseudograph’s Atom (Vertex) Adjacency Matrix”
Local (Atom-type) Linear Indices of the “Molecular Pseudograph’s Atom (Vertex) Adjacency Matrix”
Methods
Data Set for QSPR/QSAR studies
Computational Methods: TOMOCOMD-CARDD Approach
- Draw the molecular pseudograph for each molecule of the data set, using the software drawing mode. This procedure is performed by selection of the active atom symbol belonging to different groups of the periodic table,
- Use appropriate weights in order to differentiate the molecular atoms. In this work, we used as atomic property the Mulliken electronegativity [24] for each kind of atom,
- Compute the total and local linear indices of the molecular pseudograph’s atom adjacency matrix. They can be carried out in the software calculation mode, where you can select the atomic properties and the family descriptor previously to calculate the molecular indices. This software generates a table in which the rows correspond to the compounds and columns correspond to the total and local linear indices or other family molecular descriptors implemented in this program,
- Find a QSPR/QSAR equation by using mathematical techniques, such as multilinear regression analysis (MRA), Neural Networks (NN), Linear Discrimination Analysis (LDA), and so on. That is to say, we can find a quantitative relation between a property P and the linear indices having, for instance, the following appearance,where P is the measurement of the property, fk(x) is the kth total linear indices, and the ak’s are the coefficients obtained by the linear regression analysis.P = a0f0(x) + a1f1(x) + a2f2(x) +….+ akfk(x) + c
- Test the robustness and predictive power of the QSPR/QSAR equation by using internal and external cross-validation techniques,
- Develop a structural interpretation of obtained QSAR/QSPR model using total and local (atom and atom-type) linear indices as molecular descriptors.
Statistical Analysis
Results and Discussion
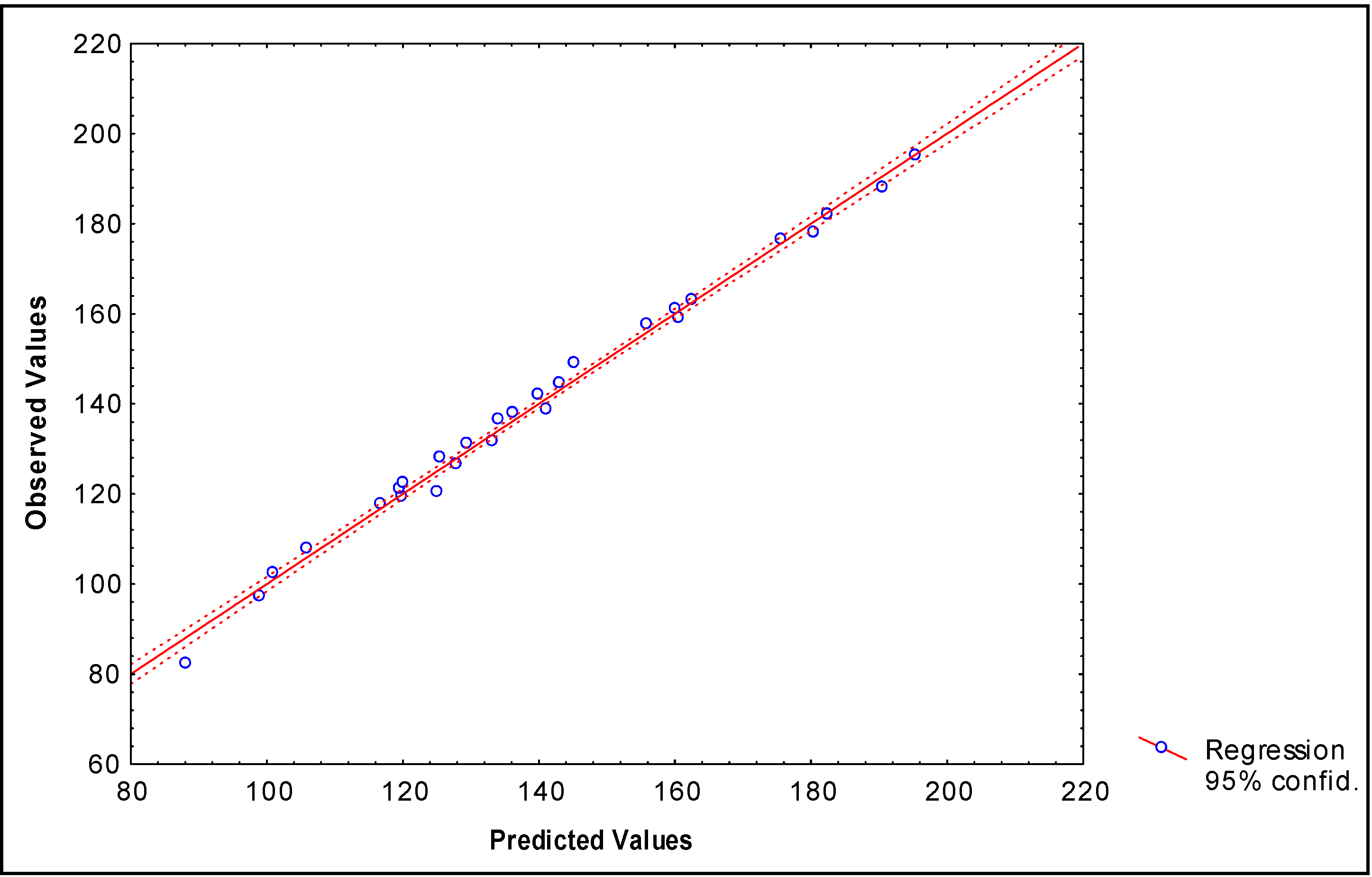
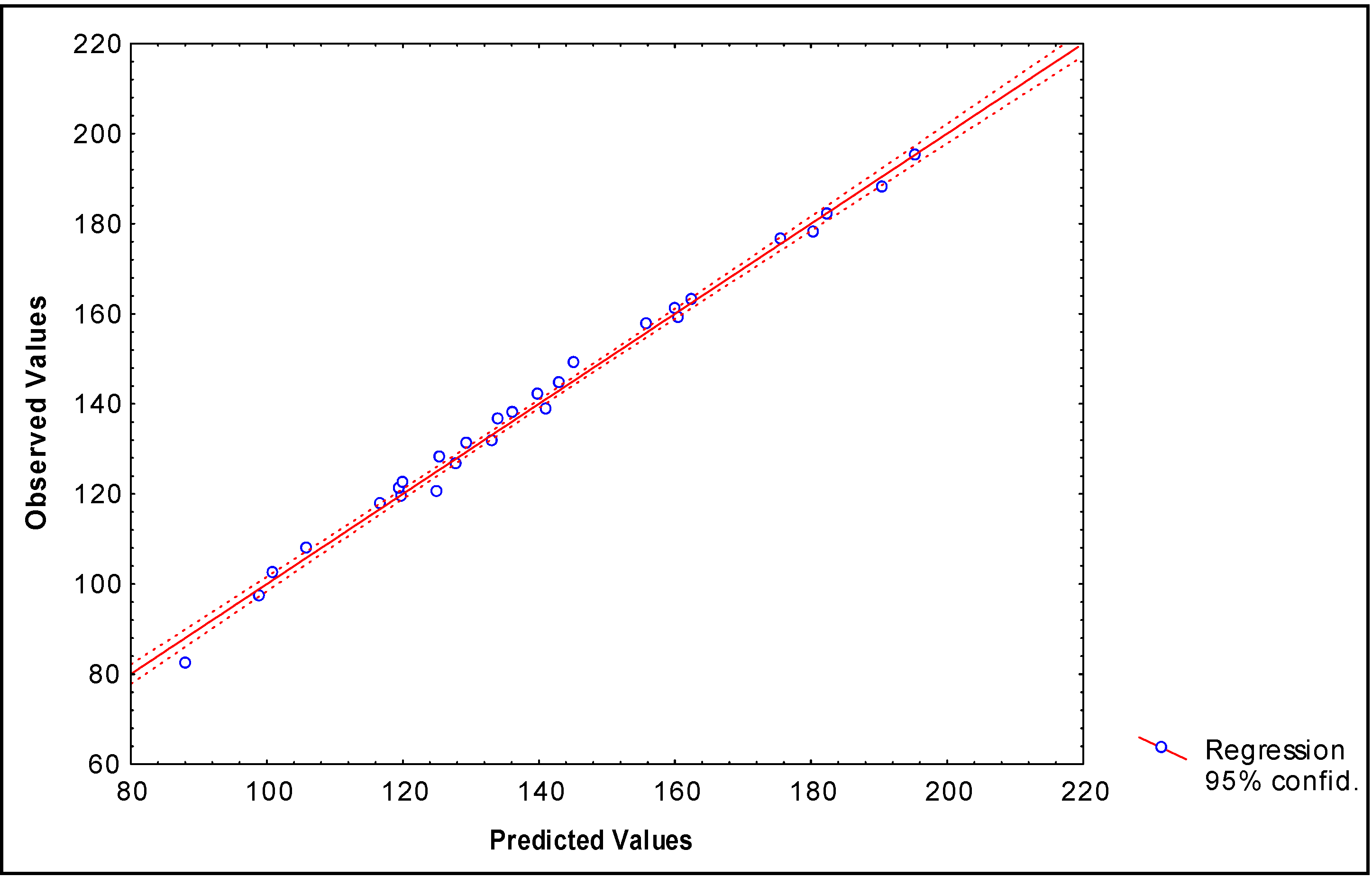
Describing boiling points of 28 alkyl alcohols
N = 28 R = 0.992 R² = 0.984 s = 3.78 q2 = 0.981 scv = 3.91 F (2,25) = 748.57
-0.024(±0.003) f7(x) -0.9526(±0.118) f HL4(xO)
N = 28 R = 0.997 R²= 0.9934 s = 2.48 q2 = 0.990 scv = 2.79 F (4,23) = 871.96
| Alcohol-R | Bp (°C)a | Predicted b Bp (°C) | Predicted c Bp (°C) | Predictedd Bp (°C) | Predictede Bp (°C) |
|---|---|---|---|---|---|
| (CH3)2CH- | 82.3 | 85.4 | 88,2 | 82.9 | 91.1 |
| CH3CH2CH2- | 97.2 | 97.5 | 99,0 | 96.0 | 97.4 |
| CH3(CH2)3- | 117.7 | 117.1 | 116,8 | 115.2 | 113.6 |
| CH3CH(CH3)CH2- | 107.8 | 107.7 | 106,0 | 108.0 | 109.0 |
| CH3CH2C(CH3)2- | 102.4 | 103.0 | 101,0 | 105.4 | 112.4 |
| CH3CH2CH2CH(CH3)- | 119.3 | 124.6 | 119,9 | 114.4 | 120.3 |
| CH3CH(CH3)CH2CH2- | 131.1 | 127.3 | 129,5 | 134.5 | 127.4 |
| CH3CH2CH(CH3)CH2- | 128.0 | 127.3 | 125,6 | 127.3 | 125.2 |
| CH3(CH2)4- | 137.9 | 136.7 | 136,3 | 134.3 | 131.8 |
| CH3C(CH3)2CH(CH3)- | 120.4 | 116.0 | 125,2 | 129.3 | 123.0 |
| CH3(CH2)2C(CH3)2- | 121.1 | 122.6 | 119,6 | 124.9 | 128.9 |
| (CH3CH2)2C(CH3)- | 122.4 | 122.6 | 120,2 | 121.9 | 126.3 |
| CH3CH2C(CH3)2CH2- | 136.5 | 128.1 | 134,2 | 142.5 | 138.4 |
| CH3CH(CH3)CH2CH(CH3)- | 131.6 | 134.8 | 133,3 | 133.9 | 133.4 |
| CH3CH(CH3)CH(CH3CH2)- | 126.5 | 134.8 | 128,0 | 121.9 | 128.7 |
| CH3CH(CH3)CH(CH3)CH2- | 144.5 | 137.5 | 143,1 | 146.7 | 138.3 |
| CH3CH2CH2CH(CH3)CH2- | 149.0 | 146.9 | 145,3 | 146.4 | 143.4 |
| CH3(CH2)5- | 157.6 | 156.3 | 155,9 | 153.4 | 169.8 |
| (CH3CH(CH3))2CH- | 138.7 | 145.0 | 141,2 | 136.4 | 139.0 |
| CH3CH(CH3)CH2CH(CH3)CH2- | 159.0 | 157.1 | 160,7 | 165.5 | 157.7 |
| (CH3CH2)3C- | 142.0 | 142.2 | 140,0 | 138.6 | 138.5 |
| CH3(CH2)6- | 176.4 | 175.9 | 175,7 | 172.5 | 172.2 |
| (CH3CH2CH2)2(CH3)C- | 161.0 | 161.8 | 160,1 | 160.9 | 161.3 |
| (CH3(CH2)3)(CH3CH2)(CH3)C- | 163.0 | 161.8 | 162,6 | 160.5 | 162.7 |
| CH3CH(CH3)CH2(CH2)4- | 188.0 | 186.1 | 190,6 | 191.6 | 188.3 |
| CH3(CH2)7- | 195.1 | 195.5 | 195,5 | 191.6 | 193.0 |
| CH3(CH2)5C(CH3)2- | 178.0 | 183.3 | 180,5 | 182.2 | 188.4 |
| (CH3CH2CH2)2(CH3CH2)C- | 182.0 | 181.4 | 182,5 | 177.6 | 177.0 |
+0.0018µ8(C–O)
N = 28 R2 = 0.982 s = 4.2 F = 23.8
-22.413(±3.85)S(-OH)
N = 28 R2 = 0.926 s = 5.8 F = 204
Modeling specific rate constants (log k) of 34 2-furylethylenes derivatives
 | |||||||
|---|---|---|---|---|---|---|---|
| no. | R1 | R2 | R3 | no. | R1 | R2 | R3 |
| 1 | H | NO2 | COOCH3 | 18 | NO2 | H | CONHCH(CH3)C2H5 |
| 2 | CH3 | NO2 | COOCH3 | 19 | NO2 | H | CONHC(CH3)3 |
| 3 | Br | NO2 | COOCH3 | 20 | NO2 | H | CONHCH2C(CH3)3 |
| 4 | I | NO2 | COOCH3 | 21 | NO2 | H | COOCH3 |
| 5 | COOCH3 | NO2 | COOCH3 | 22 | NO2 | H | COOC2H5 |
| 6 | NO2 | NO2 | COOCH3 | 23 | NO2 | H | COO(CH2)2CH3 |
| 7 | NO2 | COOC2H5 | COOC2H5 | 24 | NO2 | H | COOCH(CH3)2 |
| 8 | NO2 | H | NO2 | 25 | NO2 | H | COO(CH2)3CH3 |
| 9 | H | H | NO2 | 26 | NO2 | H | COOCH2CH(CH3)2 |
| 10 | NO2 | H | CONH2 | 27 | NO2 | H | COOCH(CH3)C2H5 |
| 11 | NO2 | H | CONHCH3 | 28 | NO2 | H | COOC(CH3)3 |
| 12 | NO2 | H | CON(CH3)2 | 29 | NO2 | H | COO(CH2)4CH3 |
| 13 | NO2 | H | CONHC2H5 | 30 | NO2 | H | Br |
| 14 | NO2 | H | CONH(CH2)2CH3 | 31 | NO2 | H | CN |
| 15 | NO2 | H | CONHCH(CH3)2 | 32 | NO2 | H | OCH3 |
| 16 | NO2 | H | CONH(CH2)3CH3 | 33 | NO2 | H | H |
| 17 | NO2 | H | CONHCH2CH(CH3)2 | 34 | NO2 | CN | COOCH3 |
| Novel R1,R2-Substituted 2-Furylethylenes (R3 = NO2) used as external test set to assess the predictive power of the classification model for antibacterial activity | |||||||
| 1 | Br | Br | NO2 | 6 | H | I | NO2 |
| 2 | I | I | NO2 | 7 | H | CH3 | NO2 |
| 3 | Br | H | NO2 | 8 | Br | CH3 | NO2 |
| 4 | H | Br | NO2 | 9 | I | CH3 | NO2 |
| 5 | I | H | NO2 | ||||
| Number | Experimental | TIs | Total moments | QC | Local moments | Linear indices |
|---|---|---|---|---|---|---|
| 1 | 6.591 | 7.532 | 6.281 | 6.780 | 6.679 | 6.441 |
| 2 | 6.518 | 6.983 | 5.583 | 6.646 | 6.563 | 6.540 |
| 3 | 6.914 | 6.352 | 6.893 | 7.021 | 6.979 | 7.075 |
| 4 | 6.982 | 6.997 | 6.554 | 6.935 | 7.069 | 7.015 |
| 5 | 7.176 | 6.750 | 7.390 | 7.137 | 7.291 | 7.188 |
| 6 | 7.602 | 6.685 | 7.410 | 7.466 | 7.572 | 7.589 |
| 7 | 5.255 | 5.533 | 5.002 | 5.212 | 4.934 | 5.063 |
| 8 | 6.763 | 4.965 | 5.962 | 6.737 | 6.662 | 6.353 |
| 9 | 5.623 | 4.997 | 4.839 | 5.454 | 5.599 | 5.292 |
| 10 | 3.813 | 4.831 | 4.455 | 3.695 | 4.318 | 4.468 |
| 11 | 3.840 | 3.636 | 4.876 | 3.616 | 4.034 | 4.005 |
| 12 | 3.874 | 4.162 | 2.911 | 3.537 | 3.448 | 3.610 |
| 13 | 3.825 | 3.916 | 4.255 | 3.562 | 3.875 | 3.722 |
| 14 | 3.623 | 3.798 | 4.032 | 3.550 | 3.841 | 3.698 |
| 15 | 3.751 | 3.528 | 3.255 | 3.522 | 3.721 | 3.461 |
| 16 | 3.784 | 3.114 | 3.437 | 3.566 | 3.842 | 3.693 |
| 17 | 3.697 | 4.175 | 3.468 | 3.570 | 3.826 | 3.684 |
| 18 | 3.705 | 3.934 | 3.157 | 3.541 | 3.711 | 3.443 |
| 19 | 3.697 | 3.264 | 3.686 | 3.482 | 3.798 | 3.227 |
| 20 | 3.650 | 3.526 | 4.269 | 3.581 | 3.851 | 3.683 |
| 21 | 4.000 | 3.693 | 5.184 | 3.985 | 3.735 | 4.259 |
| 22 | 3.920 | 3.974 | 4.202 | 3.920 | 3.617 | 3.993 |
| 23 | 3.790 | 3.856 | 4.409 | 3.926 | 3.596 | 3.970 |
| 24 | 3.763 | 3.869 | 3.438 | 3.849 | 3.552 | 3.749 |
| 25 | 3.623 | 3.172 | 3.893 | 3.921 | 3.600 | 3.965 |
| 26 | 3.650 | 4.233 | 4.178 | 3.917 | 3.610 | 3.957 |
| 27 | 3.592 | 4.193 | 3.613 | 3.873 | 3.566 | 3.732 |
| 28 | 3.584 | 4.059 | 3.596 | 3.819 | 3.881 | 3.532 |
| 29 | 3.590 | 3.478 | 3.204 | 3.918 | 3.600 | 3.968 |
| 30 | 2.987 | 3.463 | 3.534 | 3.281 | 2.785 | 2.997 |
| 31 | 3.273 | 3.070 | 3.779 | 4.089 | 3.651 | 3.370 |
| 32 | 2.140 | 3.070 | 2.794 | 2.287 | 2.751 | 1.941 |
| 33 | 3.553 | 3.780 | 2.716 | 3.338 | 3.485 | 3.558 |
| 34 | 5.557 | 5.114 | 5.474 | 4.969 | 5.524 | 5.465 |
-0.00107(±8.92x10-5)fH8(xC2-C6) +0.225923(±0.02621)fH4(xC2-C6)
+3.85x10-6(±4.53x10-7)f12(xC2-C6) +0.05(±0.0076)fH2(xC6-C7)
N = 34 R = 0.986 R² = 0.973 s = 0.26 q2 = 0.948 scv = 0.33 F(6,27) = 161.22
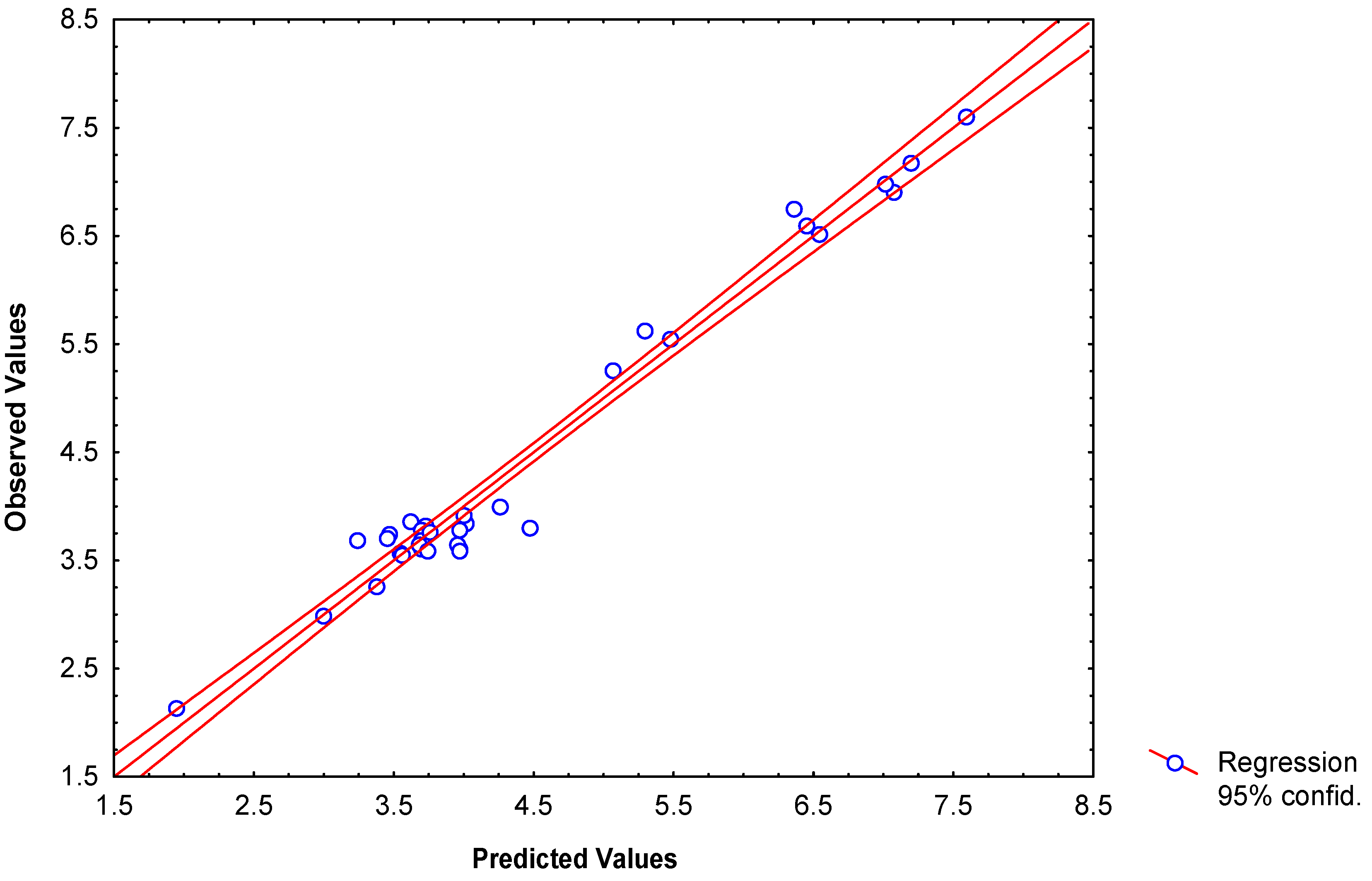
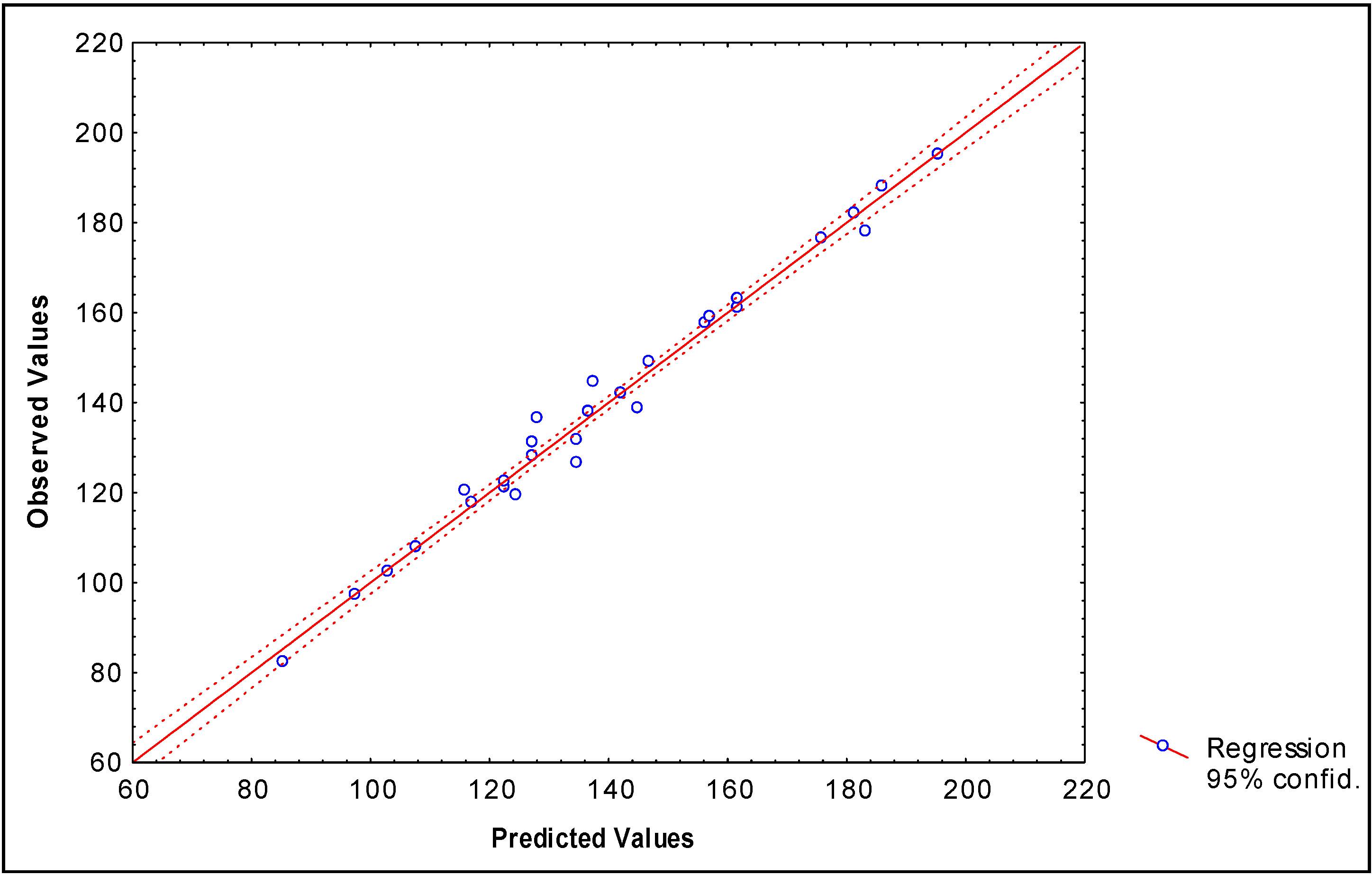
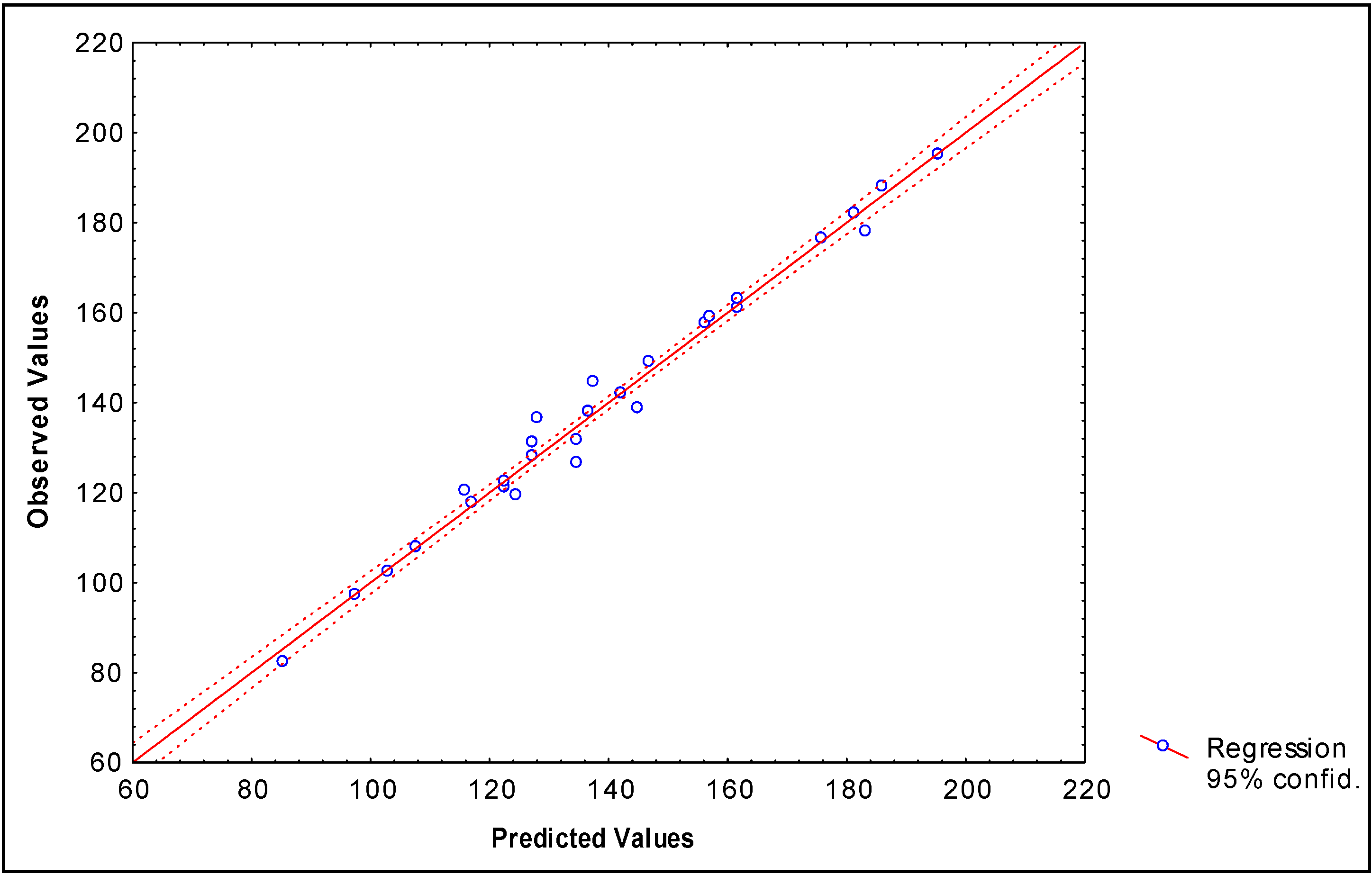
Modeling partition coefficients (log P) of 34 2-furylethylenes derivatives
-0.1576(±0.012)fHL2(xE) +0.00915(±0.0022) fHL3(xE) -0.069(±0.007)fHL2(xE-H)
+0.0026(±0.0007)fHL3(xC6-C7) +3.511x10-6(±6.23x10-6)fHL3(xC6-C7)
N = 34 R = 0.984 R² = 0.968 s = 0.143 q2 = 0.938 scv = 0.176 F(7,26) = 113.38
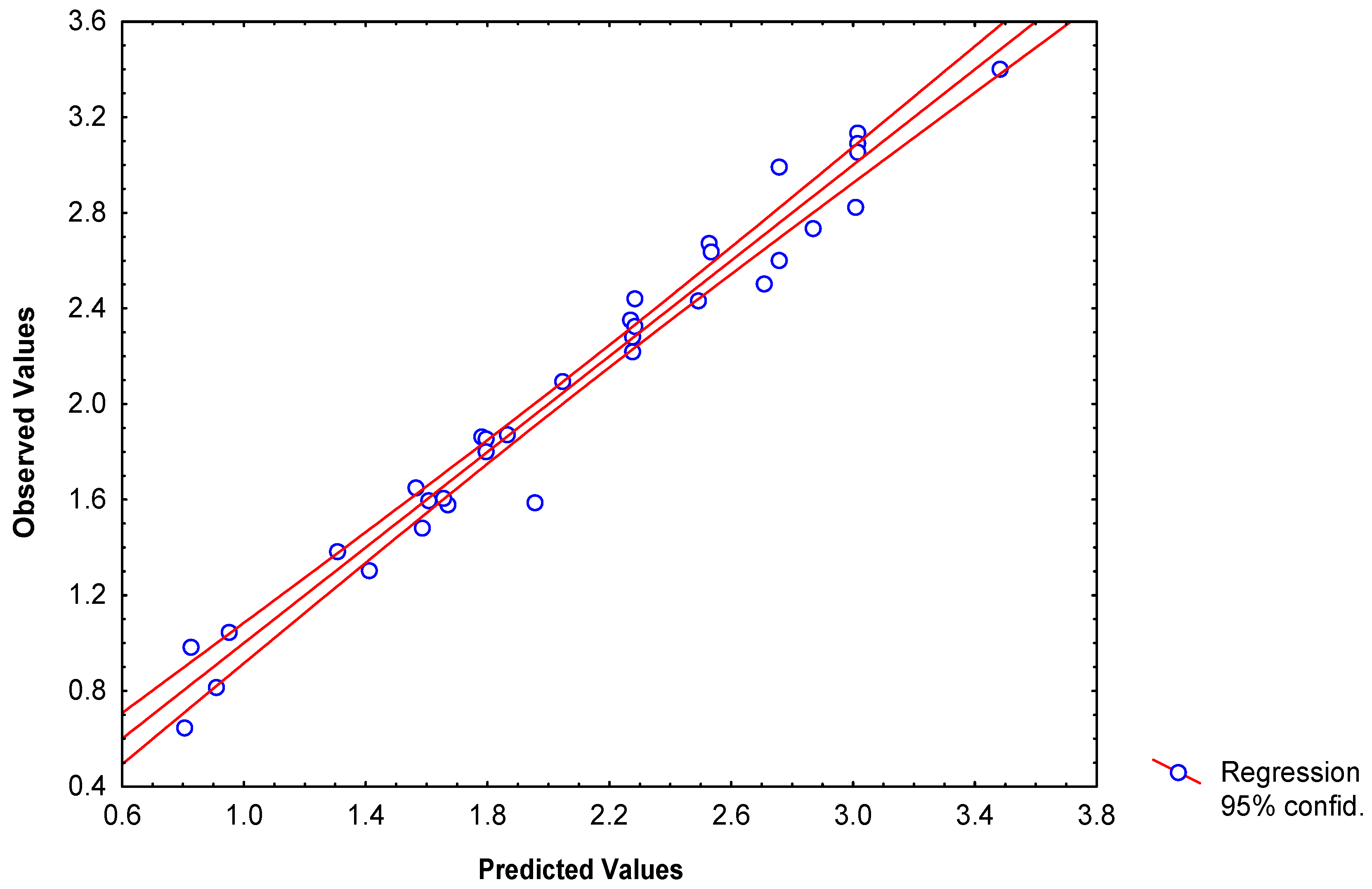
Classification of 34 2-furylethylene derivatives as antibacterial
N = 34 λ = 0.304 D2 = 9.44 F(3,30) = 22.862 p<0.0000
| no. | Obsd.a | Pred.b | Res.c | Res-CVd | topol.e | topog.f | QCg |
|---|---|---|---|---|---|---|---|
| 1 | 1.879 | 1.86 | 0.02 | 0.02 | 1.894 | 1.955 | 1.836 |
| 2 | 2.439 | 2.49 | -0.05 | -0.07 | 2.482 | 2.398 | 2.239 |
| 3 | 2.739 | 2.86 | -0.13 | -0.20 | 2.753 | 2.748 | 2.405 |
| 4 | 2.999 | 2.75 | 0.25 | 0.39 | 2.905 | 2.898 | 2.510 |
| 5 | 1.869 | 1.78 | 0.09 | 0.13 | 1.763 | 1.930 | 1.976 |
| 6 | 1.599 | 1.60 | 0.00 | -0.01 | 1.619 | 1.550 | 1.679 |
| 7 | 2.504 | 2.71 | -0.20 | -0.27 | 2.703 | 2.640 | 2.706 |
| 8 | 1.303 | 1.41 | -0.11 | -0.19 | 1.191 | 1.338 | 1.456 |
| 9 | 1.583 | 1.67 | -0.09 | -0.12 | 1.453 | 1.783 | 1.583 |
| 10 | 0.649 | 0.80 | -0.15 | -0.32 | 0.433 | 0.300 | 0.180 |
| 11 | 0.984 | 0.82 | 0.16 | 0.20 | 0.999 | 1.091 | 1.076 |
| 12 | 0.819 | 0.91 | -0.09 | -0.17 | 1.160 | 0.870 | 2.149 |
| 13 | 1.386 | 1.31 | 0.08 | 0.09 | 1.583 | 1.423 | 1.482 |
| 14 | 1.860 | 1.79 | 0.07 | 0.08 | 2.311 | 1.941 | 1.858 |
| 15 | 1.803 | 1.79 | 0.01 | 0.01 | 1.966 | 2.084 | 1.906 |
| 16 | 2.356 | 2.26 | 0.09 | 0.10 | 2.168 | 2.332 | 2.240 |
| 17 | 2.225 | 2.27 | -0.05 | -0.05 | 2.493 | 2.526 | 2.241 |
| 18 | 2.284 | 2.27 | 0.01 | 0.01 | 2.384 | 2.383 | 2.277 |
| 19 | 2.333 | 2.28 | 0.06 | 0.07 | 2.316 | 2.316 | 2.346 |
| 20 | 2.605 | 2.75 | -0.15 | -0.18 | 2.382 | 2.575 | 2.618 |
| 21 | 1.652 | 1.56 | 0.09 | 0.10 | 1.347 | 1.585 | 1.830 |
| 22 | 2.098 | 2.05 | 0.05 | 0.06 | 1.984 | 1.947 | 2.126 |
| 23 | 2.673 | 2.53 | 0.15 | 0.16 | 2.733 | 2.459 | 2.504 |
| 24 | 2.641 | 2.53 | 0.11 | 0.12 | 2.484 | 2.666 | 2.592 |
| 25 | 2.827 | 3.00 | -0.18 | -0.19 | 2.726 | 2.837 | 2.902 |
| 26 | 3.135 | 3.01 | 0.13 | 0.14 | 3.052 | 3.034 | 2.902 |
| 27 | 3.091 | 3.01 | 0.08 | 0.09 | 3.018 | 2.952 | 2.943 |
| 28 | 3.060 | 3.01 | 0.05 | 0.05 | 2.994 | 3.002 | 3.029 |
| 29 | 3.404 | 3.48 | -0.07 | -0.09 | 3.227 | 3.252 | 3.266 |
| 30 | 2.447 | 2.28 | 0.17 | 0.24 | 2.510 | 2.469 | 2.132 |
| 31 | 1.050 | 0.95 | 0.10 | 0.24 | 1.365 | 1.258 | 1.344 |
| 32 | 1.591 | 1.95 | -0.36 | -0.41 | 1.510 | 1.500 | 1.711 |
| 33 | 1.611 | 1.65 | -0.04 | -0.07 | 1.738 | 1.515 | 1.590 |
| 34 | 1.488 | 1.59 | -0.10 | -0.27 | 1.309 | 1.424 | 1.504 |
| Compd. | Obsd.[39] | Linear Indices | 2D Conn. [39] | 3D Conn. [39] | Quantum [39] | ||||
|---|---|---|---|---|---|---|---|---|---|
| Class. | Prob. | Class. | Prob. | Class. | Prob. | Class. | Prob. | ||
| 1 | + | + | 99.99 | + | 95.43 | + | 99.49 | + | 99.72 |
| 2 | + | + | 99.97 | + | 91.67 | + | 95.83 | + | 99.86 |
| 3 | + | + | 100.00 | + | 84.95 | + | 96.22 | + | 98.31 |
| 4 | + | + | 100.00 | + | 79.65 | + | 95.78 | + | 97.67 |
| 5 | + | + | 99.58 | + | 99.72 | + | 99.63 | + | 99.66 |
| 6 | + | + | 99.96 | + | 99.85 | + | 99.98 | + | 98.91 |
| 7 | + | + | 88.62 | + | 94.29 | + | 91.77 | + | 98.57 |
| 8 | + | + | 90.32 | + | 74.81 | + | 57.81 | + | 92.50 |
| 9 | + | + | 98.47 | - | 9.86 | - | 8.11 | + | 77.08 |
| 10 | + | + | 99.82 | + | 99.13 | + | 99.28 | - | 32.29 |
| 11 | + | + | 85.96 | + | 88.24 | + | 57.28 | - | 9.46 |
| 12 | + | - | 4.58 | + | 66.00 | + | 86.94 | - | 4.26 |
| 13 | + | + | 50.52 | + | 57.89 | + | 71.56 | - | 12.79 |
| 14 | - | - | 14.55 | - | 6.25 | - | 46.43 | - | 12.96 |
| 15 | - | - | 14.55 | - | 28.14 | - | 36.15 | - | 8.72 |
| 16 | - | - | 2.76 | - | 0.92 | - | 1.10 | - | 11.78 |
| 17 | - | - | 2.76 | - | 2.35 | - | 6.19 | - | 11.05 |
| 18 | - | - | 2.76 | - | 37.62 | - | 4.56 | - | 9.96 |
| 19 | - | - | 2.76 | - | 8.96 | - | 2.97 | - | 9.96 |
| 20 | - | - | 0.47 | - | 1.14 | - | 0.08 | - | 9.75 |
| 21 | - | - | 49.07 | + | 55.73 | + | 88.95 | - | 8.59 |
| 22 | - | - | 13.84 | - | 22.77 | - | 18.96 | - | 7.60 |
| 23 | - | - | 2.61 | - | 1.36 | - | 8.23 | - | 7.66 |
| 24 | - | - | 2.61 | - | 7.01 | - | 4.83 | - | 6.44 |
| 25 | - | - | 0.44 | - | 0.19 | - | 0.09 | - | 8.56 |
| 26 | - | - | 0.44 | - | 0.33 | - | 0.58 | - | 8.49 |
| 27 | - | - | 0.44 | - | 7.59 | - | 0.43 | - | 7.94 |
| 28 | - | - | 0.44 | - | 1.41 | - | 0.27 | - | 7.02 |
| 29 | - | - | 0.07 | - | 0.02 | - | 0.04 | - | 7.21 |
| 30 | - | - | 0.55 | - | 4.65 | - | 7.56 | - | 0.32 |
| 31 | - | + | 52.57 | - | 29.58 | - | 37.61 | - | 3.04 |
| 32 | - | - | 1.05 | - | 23.67 | - | 14.96 | - | 5.30 |
| 33 | - | - | 0.33 | + | 58.87 | - | 14.08 | - | 0.42 |
| 34 | + | + | 99.69 | + | 97.13 | + | 97.50 | + | 62.36 |
| Test set | |||||||||
| 1 | + | + | 99.26 | + | 88.53 | + | 95.81 | + | 87.18 |
| 2 | + | + | 99.04 | + | 86.87 | + | 94.59 | + | 85.53 |
| 3 | + | + | 98.77 | + | 59.01 | + | 65.00 | + | 54.95 |
| 4 | + | + | 99.07 | + | 96.35 | + | 99.59 | + | 96.12 |
| 5 | + | + | 98.67 | NC | 50.07 | + | 53.00 | NC | 50.01 |
| 6 | + | + | 98.89 | + | 96.72 | + | 99.51 | + | 97.68 |
| 7 | + | + | 99.99 | + | 95.92 | + | 99.62 | + | 94.76 |
| 8 | + | + | 99.99 | + | 86.38 | + | 96.06 | + | 84.27 |
| 9 | + | + | 99.99 | + | 81.75 | + | 95.74 | + | 79.29 |
| index | n | R2 | s | q2 | sCV | F |
|---|---|---|---|---|---|---|
| Boiling Point of 28 Alkyl-Alcohols | ||||||
| Linear indices (Eq. 12) | 2 | 0.984 | 3.78 | 0.981 | 3.91 | 748.57 |
| Linear indices (Eq. 13) | 4 | 0.993 | 2.48 | 0.99 | 2.79 | 871.96 |
| Local spectral moments [11] | 5 | 0.982 | 4.2 | * | * | 23.8 |
| E-State/encounter parameters [13] | 3 | 0.926 | 5.8 | * | * | 204 |
| Reactivity (log k) of 34 2-Furylethylenes | ||||||
| Linear indices | 6 | 0.973 | 0.260 | 0.948 | 0.33 | 161.2 |
| Conn. Indices [11] | 7 | 0.821 | 0.681 | * | * | 17.1 |
| Global spectral moments [11] | 7 | 0.843 | 0.655 | * | * | 18.8 |
| Local spectral moments [11] | 7 | 0.964 | 0.320 | * | * | 70.4 |
| Quantum chemical descriptors [11] | 7 | 0.968 | 0.288 | * | * | 112.2 |
| Partition Coefficient n-Octanol/Water (log P) of 34 2-Furylethylenes | ||||||
| Linear indices | 7 | 0.968 | 0.143 | 0.938 | 0.176 | 113.38 |
| Vertex and edge conn. Indices [39] | 7 | 0.939 | 0.199 | * | 0.247 | 56.9 |
| Topographic descriptors [39] | 7 | 0.964 | 0.155 | * | 0.176 | 84.6 |
| quantum chemical descriptors [39] | used the Rogers and Cammarata approach | 0.875 | 0.319 | * | 0.370 | 45.5 |
| index | n | λ | D2 | Accuracy (Training) | Accuracy (Test) | F |
| Classification of 34 2-Furylethylene Derivatives as Antibacterial | ||||||
| Linear indices | 3 | 0.30 | 9.44 | 94.12% | 100% | 22.9 |
| Vertex and edge conn. Indices [39] | 5 | 0.43 | 5.7 | 91.2% | 100% | 7.7 |
| Topographic descriptors [39] | 5 | 0.38 | 6.7 | 94.1% | 100% | 9.1 |
| quantum chemical descriptors [39] | 5 | 0.44 | 5.2 | 88.2% | 100% | 7.1 |
Concluding Remarks
Acknowledgements
References
- Julián-Ortiz, J. V. Virtual Darwinian Drug Design: QSAR Inverse Problem, Virtual Combinatorial Chemistry, and Computational Screening. Comb. Chem. High Throughput Screen. 2001, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Dean, P.M.; Lewis, R.A. (Eds.) Molecular Diversity in Drug Design; Kluwer Academic Publishers: Amsterdam, 1999.
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening: An overview. Drug Discovery Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Karelson, M. Molecular Descriptors in QSAR/ QSPR; John Wiley & Sons: New York, 2000. [Google Scholar]
- Katritzky, A. R.; Gordeeva, E. V. Traditional Topological Indexes vs Electronic, Geometrical, and Combined Molecular Descriptors in QSAR/QSPR Research. J. Chem. Inf. Comput. Sci. 1993, 3, 835. [Google Scholar] [CrossRef]
- Kier, L. B.; Hall, L. H. Molecular Connectivity in Chemistry and Drug Research; Academic Press: London, 1976. [Google Scholar]
- Balaban, A. Topological and Stereochemical Molecular Descriptors for Databases Useful in QSAR, Similarity/Dissimilarity and Drug Design. SAR QSAR Environ. Res. 1998, 8, 1–21. [Google Scholar] [CrossRef]
- Estrada, E. On the Topological Sub-Structural Molecular Design (TOSS-MODE) in QSPR/QSAR and Drug Design Research. SAR QSAR Environ. Res. 2000, 11, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Julian-Ortiz, J. V.; Gálvez, J.; Muños-Collado, C.; García- Domenech, R.; Gimeno-Cardona, C. Virtual Combinatorial Syntheses and Computational Screening of New Potential Anti-Herpes Compounds. J. Med. Chem. 1999, 42, 3308–3314. [Google Scholar]
- Estrada, E.; Uriarte, E. Recent Advances on the Role of Topological Indices in Drug Discovery Research. Curr. Med. Chem. 2001, 8, 1573–1588. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.; Molina, E. Novel Local (Fragment-Based) Topological Molecular Descriptors for QSPR/QSAR and Molecular Design. J. Mol. Graphics Mod. 2001, 20, 54–64. [Google Scholar] [CrossRef]
- Kier, L.B.; Hall, L.H. The electrotopological state: structure modeling for QSAR and data base analysis. In Topological Indices and Related Descriptors in QSAR and QSPR; Devillers, J., Balaban, A.T., Eds.; Gordon and Breach: Amsterdam, 1999; pp. 491–562. [Google Scholar]
- Kier, L.B.; Hall, L.H. MolecularStructure Description. The Electrotopological State; Academic Press: New York, 1999. [Google Scholar]
- Marrero-Ponce, Y.; Romero, V. TOMOCOMD software. Central University of Las Villas. 2002. TOMOCOMD (TOpological MOlecular COMputer Design) for Windows, version 1.0 is a preliminary experimental version; in the future a professional version will be available upon request from Y. Marrero: [email protected]; [email protected].
- Marrero-Ponce, Y. Total and Local Quadratic Indices of the Molecular Pseudograph’s Atom Adjacency Matrix: Applications to the Prediction of Physical Properties of Organic Compounds. Molecules 2003, 8, 687–726, http://www.mdpi.org. [Google Scholar]
- Marrero-Ponce, Y. Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Definition, Significance-Interpretation and Application to QSAR Analysis of Flavone Derivatives as HIV-1 Integrase Inhibitors. J. Chem. Inf. Comput. Sci. [CrossRef]
- Marrero-Ponce, Y.; Cabrera, M.A.; Romero, V.; Ofori, E.; Montero, L. A. Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs. Int. J. Mol. Sci. 2003, 4, 512–536. [Google Scholar]
- Marrero-Ponce, Y.; Cabrera, M. A.; Romero, V.; González, D. H.; Torrens, F. A New Topological Descriptors Based Model for Predicting Intestinal Epithelial Transport of Drugs in Caco-2 Cell Culture. J. Pharm. & Pharm. Sci. 2004, 7, 186–199. [Google Scholar]
- Marrero-Ponce, Y.; Nodarse, D.; González-Díaz, H.; Ramos de Armas, R.; Romero-Zaldivar, V.; Torrens, F.; Castro, E. Nucleic Acid Quadratic Indices of the “Macromolecular Graph’s Nucleotides Adjacency Matrix”. Modeling of Footprints after the Interaction of Paromomycin with the HIV-1 Ψ-RNA Packaging Region. CPS: physchem/0401004.
- Marrero-Ponce, Y.; Castillo-Garit, J.A.; Olazabal, E.; Serrano, H. S.; Morales, A.; Castañedo, N.; Ibarra-Velarde, F.; Huesca-Guillen, A.; Jorge, E.; del Valle, A.; Torrens, F.; Castro, E.A. TOMOCOMD-CARDD, a Novel Approach for Computer-Aided “Rational” Drug Design: I. Theoretical and Experimental Assessment of a Promising Method for Computational Screening and in silico Design of New Anthelmintic Compounds. In J. Comput. Aided Mol. Des.; Submitted for publication.
- Marrero-Ponce, Y.; Iyarreta-Veitía, M.; Montero-Torres, A.; Romero-Zaldivar, C.; Brandt, C. A.; Ávila, P. E.; Kirchgatter, K. A Novel Approach for Computer-Aided “Rational” Drug Design: Theoretical and Experimental Assessment of a Promising Method for Virtual Screening and in silico Design of New Antimalarial Compounds. In J. Med. Chem.; Submitted for publication.
- Marrero-Ponce, Y.; González-Díaz, H.; Romero-Zaldivar, V.; Torrens, F.; Castro, E. A. 3D-Chiral Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix” and their Application to Central Chirality Codification: Classification of ACE Inhibitors and Prediction of σ-Receptor Antagonist Activities. Bioorg. Med. Chem. 2004, 12, 5331–5342. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y. Total and Local (Atom and Atom-Type) Molecular Quadratic Indices: Significance-Interpretation, Comparison to Other Molecular Descriptors and QSPR/QSAR Applications. Bioorg. Med. Chem. [CrossRef]
- Cotton, F. A. Advanced Inorganic Chemistry; Ed. Revolucionaria: Havana (Cuba), 1970; p. 103. [Google Scholar]
- Wang, R.; Gao, Y.; Lai, L. Calculating Partition Coefficient by Atom-Additive Method. Perspect. Drug Discov. Des. 2000, 19, 47–66. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A. K.; Crippen, G. M. Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci. 1987, 27, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Millar, K. J. Additivity Methods in Molecular Polarizability. J. Am. Chem. Soc. 1990, 112, 8533–8542. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsilli, M. A New Model for Calculating Atomic Charge in Molecules. Tetrahedron Lett. 1978, 34, 3181–3184. [Google Scholar] [CrossRef]
- Browder, A. Mathematical Analysis. An Introduction; Springer-Verlag: New York, 1996; pp. 176–296. [Google Scholar]
- Axler, S. Linear Algebra Done Right; Springer-Verlag: New York, 1996; pp. 37–70. [Google Scholar]
- Ross, K. A.; Wright, C. R. B. Matemáticas discretas; Prentice Hall Hispanoamericana: Mexico D.F., 1990. [Google Scholar]
- Maltsev, A. I. Fundamentos del Álgebra Lineal; Mir: Moscow, 1976; pp. 68–262. [Google Scholar]
- Harary, F. Graph Theory; Addison-Wesley: Reading, MA, 1969; p. 10. [Google Scholar]
- Chartrand, G. Graph as Mathematical Models; Prindle, Weber & Schmidt: Boston, MA, 1977; p. 22. [Google Scholar]
- Wilson, R.J. Introduction to Graph Theory; Oliver & Boyd: Edinburgh, 1972; p. 10. [Google Scholar]
- Trinajstic, N. Chemical Graph TheoryCRC Press: Boca Raton, FL, 1992, 2nd edition; pp. 6–7. [Google Scholar]
- Balaz, S.; Sturdik, E.; Rosenberg, M.; Augustin, J.; Skara, B. Kinetics of Drug Activities as Influenced by Their Physicochemical Properties: Antibacterial Effects of Alkylating 2-furylethylenes. J. Theor. Biol. 1988, 131, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.; Molina, E. 3D Conectivity Indices in QSPR/QSAR Studies. J. Chem. Inf. Comput. Sci. 2001, 41, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Dore, J. Ch.; Viel, C. Antitumoral Chemoterapy. X. Cytotoxic and Antitumoral Activity of β-Nitrostyrenes and Nitrovinyl Derivatives. Farmaco. 1975, 30, 81–109. [Google Scholar]
- Sturdik, E.; Drobnica, L.; Balaz, S. Reaction of 2-Furylethylenes with Thiols in Vivo. Coll. Czch. Chem. Comm. 1985, 50, 470–480. [Google Scholar] [CrossRef]
- Blondeau, J. M.; Castañedo, N.; Gonzalez, O.; Medina, R.; Silveira, E. In Vitro Evaluation of G-1: A Novel Antimicrobial Compound. Antimicrob. Agents Chemother. 1999, 11, 1663–1669. [Google Scholar]
- Castañedo, N.; Goizueta, R.; Perez, J.; Gonzalez, J.; Silveira, E.; Cuesta, M.; Martinez, A.; Lugo, E.; Estrada, E.; Carta, A.; Navia, O.; Delgado, M. Cuban Pat. 22446, 1994; Can. Pat. 2,147,594, 1999.
- STATISTICA ver. 5.5, Statsoft, Inc. 1999.
- Wold, S.; Erikson, L. Statistical Validation of QSAR Results. Validation Tools. In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH Publishers: New York, 1995; pp. 309–318. [Google Scholar]
- Belsey, D. A.; Kuh, E.; Welsch, R. E. Regression Diagnostics; Wiley: New York, 1980. [Google Scholar]
- Cronin, M. T. D.; Schultz, T. W. Pitfalls in QSAR. J. Mol. Struct. (Theochem). 2003, 622, 39–51. [Google Scholar] [CrossRef]
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ponce, Y.M.; Garit, J.A.C.; Torrens, F.; Zaldivar, V.R.; Castro, E.A. Atom, Atom-Type, and Total Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to QSPR/QSAR Studies of Organic Compounds. Molecules 2004, 9, 1100-1123. https://doi.org/10.3390/91201100
Ponce YM, Garit JAC, Torrens F, Zaldivar VR, Castro EA. Atom, Atom-Type, and Total Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to QSPR/QSAR Studies of Organic Compounds. Molecules. 2004; 9(12):1100-1123. https://doi.org/10.3390/91201100
Chicago/Turabian StylePonce, Yovani Marrero, Juan Alberto Castillo Garit, Francisco Torrens, Vicente Romero Zaldivar, and Eduardo A. Castro. 2004. "Atom, Atom-Type, and Total Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to QSPR/QSAR Studies of Organic Compounds" Molecules 9, no. 12: 1100-1123. https://doi.org/10.3390/91201100