Protein Quadratic Indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”. 1. Prediction of Arc Repressor Alanine-mutant’s Stability

Abstract
:Introduction
Computational Methods
Arc Dimer Structure and Melting Temperature of a Complete Set of A-Substitution Mutants
- i)
- Between side chain in the same subunit (R16-D20, D20-R23, N29-E36, E36-R31, E36-R40, E43-K46, E43-K47) and; those between side chains in different subunits (E28-R50, R40-S44, R40-F48).
- ii)
- Between a side chain and main-chain atom intersubunit (W14-N34, N34-R13) and; those between a side chain and main-chain atom intrasubunits (E17-E17, S32-S35, S44-R40).

{kind=link}
{kind=link}
| Protein | Classb | P% (P)c | P% (H)c | Scored | tm(Obs)e | tm(Pred)f | Resg | tm(Pred)h | Resg |
|---|---|---|---|---|---|---|---|---|---|
| 1PA8-st6a | H | 4.31 | 95.69 | 1.47 | 74.1 | (55.1) i | 19.0 | 56.86 | 17.2 |
| 2SA35-st6 | H | 5.25 | 94.75 | 1.36 | 63.4 | 62.4 | 1.0 | 69.1 | -5.7 |
| *3NA34-st11 | H | 59.40 | 40.60 | -0.23 | 63.0 | 61.2 | 1.8 | 52.6 | 10.4 |
| 4NA11-st6a | H | 40.89 | 59.11 | 0.13 | 62.1 | 54.5 | 7.6 | 49.95 | 12.1 |
| 5QA39-st11 | H | 9.25 | 90.75 | 1.07 | 61.4 | 59.7 | 1.7 | 62.7 | -1.3 |
| *6GA52-st11 | H | 86.94 | 13.06 | -0.98 | 60.9 | 60.0 | 0.9 | 57.5 | 3.4 |
| 7KA6-st6a | H | 8.75 | 91.25 | 1.10 | 59.6 | 55.0 | 4.6 | 60.83 | -1.2 |
| 8RA16-st6 | H | 0.43 | 99.57 | 2.61 | 59.5 | 56.3 | 3.2 | 57.6 | 1.9 |
| 9VA25-st6 | H | 11.48 | 88.52 | 0.95 | 59.3 | 57.3 | 2.0 | 56.4 | 2.9 |
| 10MA4-st6 | H | 12.49 | 87.51 | 0.90 | 59.2 | 58.1 | 1.1 | 60.1 | -0.9 |
| 11Arc-st6a | H | 9.11 | 90.89 | 1.08 | 59 | 54.7 | 4.3 | 57.88 | 1.1 |
| 12EA27-st6 | H | 5.42 | 94.58 | 1.35 | 58.8 | 58.1 | 0.7 | 56.5 | 2.3 |
| 13KA2-st6 | H | 2.09 | 97.91 | 1.83 | 58.7 | 58.2 | 0.5 | 59.2 | -0.5 |
| 14QA9-st6 | H | 14.28 | 85.72 | 0.83 | 58.4 | 57.5 | 0.9 | 55.3 | 3.1 |
| 15GA3-st6 | H | 6.12 | 93.88 | 1.29 | 58.1 | 60.3 | -2.2 | 57.3 | 0.8 |
| 16MA1-st6a | H | 12.84 | 87.16 | 0.89 | 58 | 55.0 | 3.0 | 59.41 | -1.4 |
| *17Arc-st11 | H | 88.80 | 11.20 | -1.06 | 57.9 | 59.0 | -1.1 | 52.4 | 5.5 |
| 18SA5-st6 | H | 8.09 | 91.91 | 1.14 | 57.5 | 58.2 | -0.7 | 58.8 | -1.3 |
| 19RA13-st6 | H | 2.28 | 97.72 | 1.79 | 57.3 | 57.7 | -0.4 | 53.9 | 3.4 |
| 20KA46-st11 | H | 8.04 | 91.96 | 1.14 | 57.1 | 55.9 | 1.2 | 56.1 | 1.0 |
| 21EA17-st6a | H | 4.58 | 95.42 | 1.43 | 57 | 55.8 | 1.2 | 56.90 | 0.1 |
| 22VA18-st6 | H | 6.25 | 93.75 | 1.28 | 56.9 | 58.1 | -1.2 | 55.4 | 1.5 |
| 23RA23-st11 | H | 18.53 | 81.47 | 0.67 | 56.7 | 57.7 | -1.0 | 51.8 | 4.9 |
| 24KA24-st11 | H | 29.57 | 70.43 | 0.38 | 56.3 | 57.9 | -1.6 | 49.3 | 7.0 |
| 25EA43-st6 | H | 2.04 | 97.96 | 1.84 | 56.1 | 57.6 | -1.5 | 54.7 | 1.4 |
| 26EA28-s11a | H | 47.66 | 52.34 | 0.00 | 55.7 | 56.2 | -0.5 | 50.19 | 5.5 |
| 27MA7-st6 | H | 8.75 | 91.25 | 1.10 | 55.5 | 58.4 | -2.9 | 60.8 | -5.3 |
| 28DA20-st6 | H | 2.68 | 97.32 | 1.71 | 55.3 | 57.7 | -2.4 | 49.6 | 5.7 |
| 29IA51-st11 | P | 93.91 | 6.09 | -1.39 | 50.9 | 40.4 | 10.5 | 47.7 | 3.2 |
| 30GA49-st11a | P | 91.79 | 8.21 | -1.23 | 48.7 | 47.0 | 1.7 | 40.71 | 8.0 |
| *31LA19-st6 | P | 9.99 | 90.01 | 1.03 | 48.3 | 45.4 | 2.9 | 51.8 | -3.5 |
| 32GA30-st11 | P | 52.78 | 47.22 | -0.10 | 47.9 | 42.5 | 5.4 | 56.1 | -8.2 |
| 33RA50-st11 | P | 62.68 | 37.32 | -0.30 | 47.9 | 44.5 | 3.4 | 49.5 | -1.6 |
| *34KA47-st11 | P | 20.15 | 79.85 | 0.62 | 47.2 | 50.0 | -2.8 | 40.7 | 6.5 |
| 35PA15-st11a | P | 66.88 | 33.12 | -0.39 | 46.6 | 38.4 | 8.2 | 55.56 | -9.0 |
| 36SA44-st11 | P | 99.90 | 0.10 | -3.42 | 46.3 | 44.3 | 2.0 | 37.0 | 9.3 |
| 37NA29-st11 | P | 80.97 | 19.03 | -0.76 | 45.3 | 47.7 | -2.4 | 49.6 | -4.3 |
| 38VA33-st11 | P | 94.46 | 5.54 | -1.43 | 44.1 | 41.5 | 2.6 | 49.8 | -5.7 |
| 39EA48-st11 | P | 82.37 | 17.63 | -0.80 | 43.2 | 42.3 | 0.9 | 44.7 | -1.5 |
| 40LA12-st11 | P | 97.37 | 2.63 | -1.81 | 42.3 | 44.3 | -2.0 | 43.2 | -0.9 |
| *41FA10-st6a | P | 31.24 | 68.76 | 0.34 | 40.6 | 45.8 | -5.2 | 49.41 | -8.8 |
| 42LA21-st11 | P | 90.68 | 9.32 | -1.16 | 39.6 | 39.9 | -0.3 | 46.7 | -7.1 |
| *43RA31-st11 | P | 15.18 | 84.82 | 0.79 | 37.1 | 41.6 | -4.5 | 45.8 | -8.7 |
| 44MA42-st11 | P | 84.06 | 15.94 | -0.86 | 35.6 | 37.5 | -1.9 | 35.6 | 0.0 |
| 45SA32-st11a | P | 90.07 | 9.93 | -1.13 | 33.5 | 34.2 | -0.7 | 61.35 | -27.8 |
| 46YA38-st11 | P | 90.77 | 9.23 | -1.17 | 33.0 | 40.6 | -7.6 | 36.4 | -3.4 |
| 47WA14-st11 | P | 97.38 | 2.62 | -1.82 | 31.5 | 38.8 | -7.3 | 36.6 | -5.1 |
| 48RA40-st11 | P | 98.44 | 1.56 | -2.08 | 31.2 | 30.2 | 1.0 | 40.6 | -9.4 |
| 49VA22-st11 | P | 83.85 | 16.15 | -0.85 | <20 | ||||
| 50EA36-st11a | P | 69.58 | 30.42 | -0.45 | <20 | ||||
| 51IA37-st11 | P | 91.53 | 8.47 | -1.21 | <20 | ||||
| 52VA41-st11 | P | 95.81 | 4.19 | -1.58 | <20 | ||||
| 53FA45-st11 | P | 99.52 | 0.48 | -2.66 | <20 | ||||
Protein Quadratic Indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”
| Amino Acids | z1 | z2 | z3 | |
|---|---|---|---|---|
| Ala | A | 0.07 | -1.73 | 0.09 |
| Val | V | -2.69 | -2.53 | -1.29 |
| Leu | L | -4.19 | -1.03 | -0.98 |
| Ile | I | -4.44 | -1.68 | -1.03 |
| Pro | P | -1.22 | 0.88 | 2.23 |
| Phe | F | -4.92 | 1.30 | 0.45 |
| Trp | W | -4.75 | 3.65 | 0.85 |
| Met | M | -2.49 | -0.27 | -0.41 |
| Lys | K | 2.84 | 1.41 | -3.14 |
| Arg | R | 2.88 | 2.52 | -3.44 |
| His | H | 2.41 | 1.74 | 1.11 |
| Gly | G | 2.23 | -5.36 | 0.30 |
| Ser | S | 1.96 | -1.63 | 0.57 |
| Thr | T | 0.92 | -2.09 | -1.40 |
| Cys | C | 0.71 | -0.97 | 4.13 |
| Tyr | Y | -1.39 | 2.32 | 0.01 |
| Asn | N | 3.22 | 1.45 | 0.84 |
| Gln | Q | 2.18 | 0.53 | -1.14 |
| Asp | D | 3.64 | 1.13 | 2.36 |
| Glu | E | 3.08 | 0.39 | -0.07 |
= 1 if i = j and the amino acid i has a hydrogen bond between its side chain and
its main-chain atom
= 0 otherwise
= 1/2 kaij if vi or vj are vertices (amino-acid) contained within FR but not both
= 0 otherwise
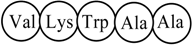 Pentapeptide Structure (sequence) 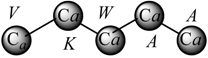 Macromolecular ‘Pseudograph’ (Gm) of the α-Carbon Atoms (Polypeptide’s backbone) Macromolecular ‘Pseudograph’ (Gm) of the α-Carbon Atoms (Polypeptide’s backbone) Amino acid residue (side chain R) Amino acid residue (side chain R)Here, we consider only covalent interaction (peptidic bond), but non-covalent interaction (hydrogen-bond and salt bridge interaction) can be taken into consideration (within a chain as well as between chains) | Macromolecular Vector: Xm = [V K W A A] ℜ5 In the definition of the Xm, as macromolecular vector, the one letter symbol of the amino acids indicates the corresponding side-chain amino-acid property, e.g., z1-values. That is to say, if we write V it means z1(V), z1-values or some amino acid property, which characterizes each side chain in the polypeptide. Therefore, if we use the canonical bases of R5, the coordinates of any vector Xm coincide with the components of that macromolecular vector [mX]t = [-2.69 2.84 -4.75 0.07 0.07] [mX]t = transposed of [mX] and it means the vector of the coordinates of Xm in the canonical basis of R5 (an 1x5 matrix) [mX]: vector of coordinates of Xm in the canonical basis of R5 (an 5x1matrix) | |||||
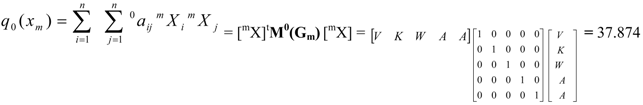 | ||||||
 | ||||||
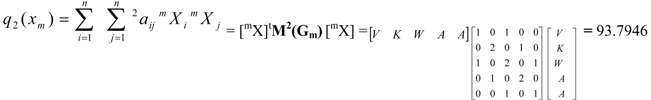 | ||||||
| Total (whole molecule) protein quadratic indices of zero, first and second order are a quadratic maps; qk(xm): ℜn→ ℜ such that, q0(V, K, W, A, A) = (V2+K2+W2+A2+A2) = 37.874 q1(V, K, W, A, A) = (2VK+KW+2WA+2AA) = -42.9144 q2(V, K, W, A, A) = (A2+V2+2K2+2W2+2A2+2WV+2AW) = 93.7946 | ||||||
If the peptide is partitioned into each (5) amino acid, the matrix Mk(Gm) can be partitioned into 5 local matrices MkL(Gm), L = 1,... 5. The kth power of the matrix M(Gm) is exactly the sum of the kth power of the local (5) matrices: 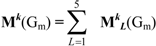 | ||||||
| The zero, first and second powers of the local (amino-acid) matrix | ||||||
 | 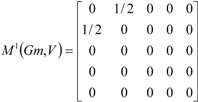 |  | ||||
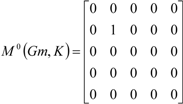 | 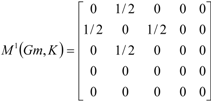 | 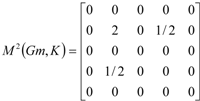 | ||||
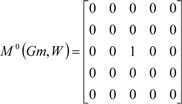 |  | 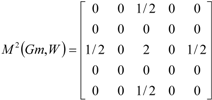 | ||||
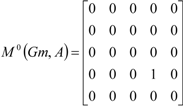 |  |  | ||||
 | 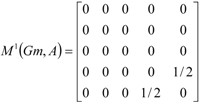 |  | ||||
and the total (whole-molecule) quadratic indices are the sum of the macromolecular quadratic indices of the 5 amino-acids, qk(xm) = 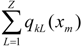 | ||||||
| Amino Acid (AA) | q0L(xm, AA) | q1L(xm, AA) | q2L(xm, AA) | q3L(xm, AA) | q4L(xm, AA) | |
| Val (V) | 7.2361 | -7.6396 | 20.0136 | -15.4675 | 52.6164 | |
| Lys (K) | 8.0656 | -21.1296 | 16.33 | -55.5504 | 41.1232 | |
| Trp (W) | 22.5625 | -13.8225 | 57.57 | -41.4675 | 172.71 | |
| Ala (A) | 0.0049 | -0.3276 | 0.2086 | -1.176 | 0.8197 | |
| Ala (A) | 0.0049 | 0.0049 | -0.3276 | 0.2086 | -1.176 | |
| Pentapeptide | 37.874 | -42.9144 | 93.7946 | -113.453 | 266.0933 | |
TOMOCOMD Software
- Draw the macromolecular pseudographs for each protein of the data set, using the software’s drawing mode. This procedure is carried out by a selection of the active aminoacid symbol belonging to ‘natural’ aminoacid code. Here, we consider only covalent interaction (peptidic bond) and hydrogen-bond interaction (within a chain as well as between chains). Afterward, we draw the mutants by changing an AA for alanine and considering that this change only affect the possibility of this region of the protein to form polar interaction (because we suppressed the hydrogen interaction if the former AA had it).
- Compute the protein quadratic indices of the “macromolecular pseudograph’s α-carbon atom adjacency matrix”. They can be performed in the software calculation mode, in which one can select the side chain properties and the family descriptor previously to calculate the molecular indices. This software generates a table in which the rows and columns correspond to the compounds and the qk(xm), respectively.
- Find a QSPR/QSAR equation by using statistical techniques, such as multilinear regression analysis (MRA), Neural Networks (NN), Linear Discrimination Analysis (LDA), and so on. That is to say, we can find a quantitative relation between a property P and the qk(xm) having, for instance, the following appearance,where P is the measurement of the property, qk(xm) [or qkL(xm)] is the kth total [or local] macromolecular quadratic indices, an the ak’s are the coefficients obtained by the statistical analysis.P = a0q0(x) + a1q1(x) + a2q2(x) +….+ akqk(x) + c
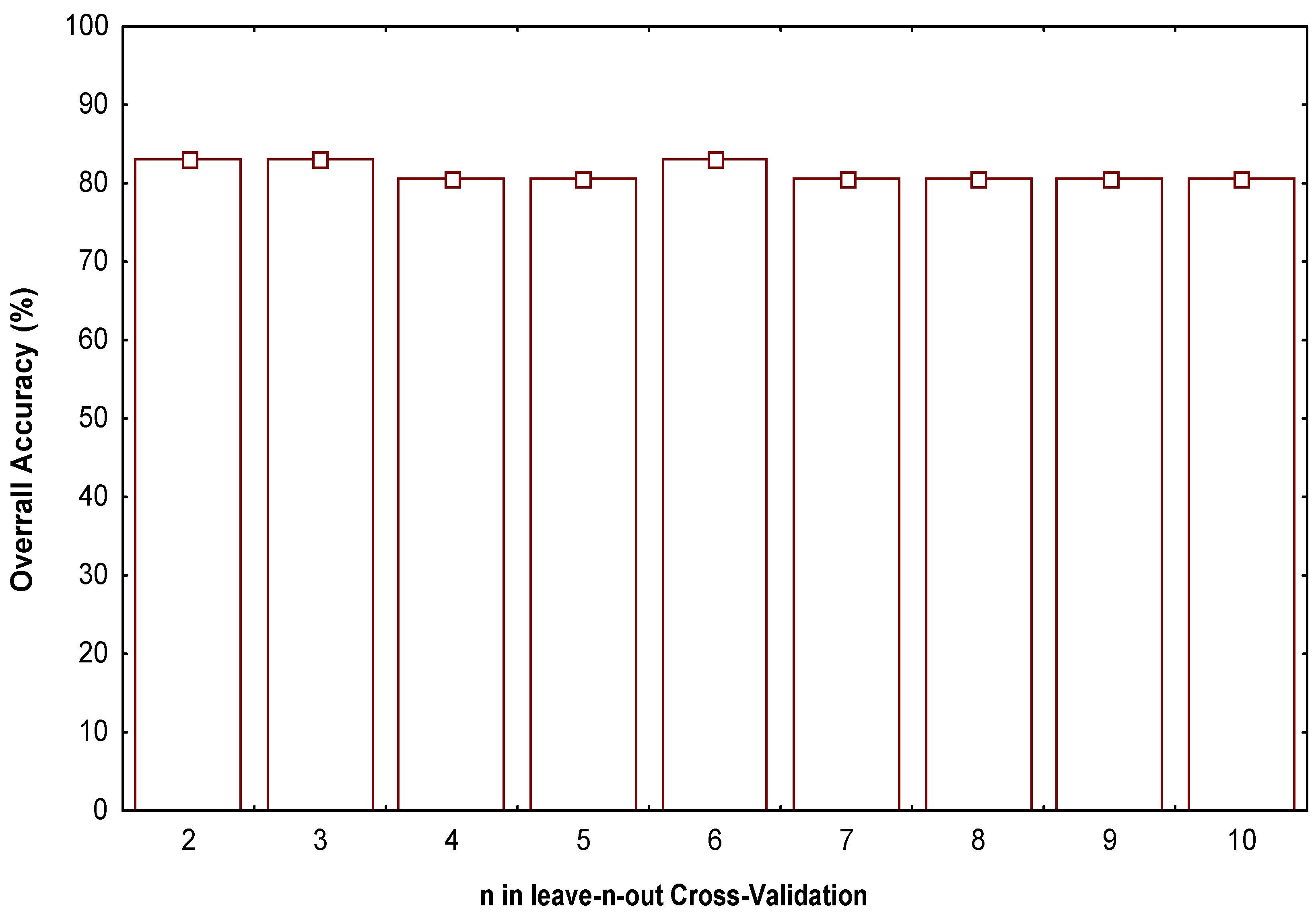
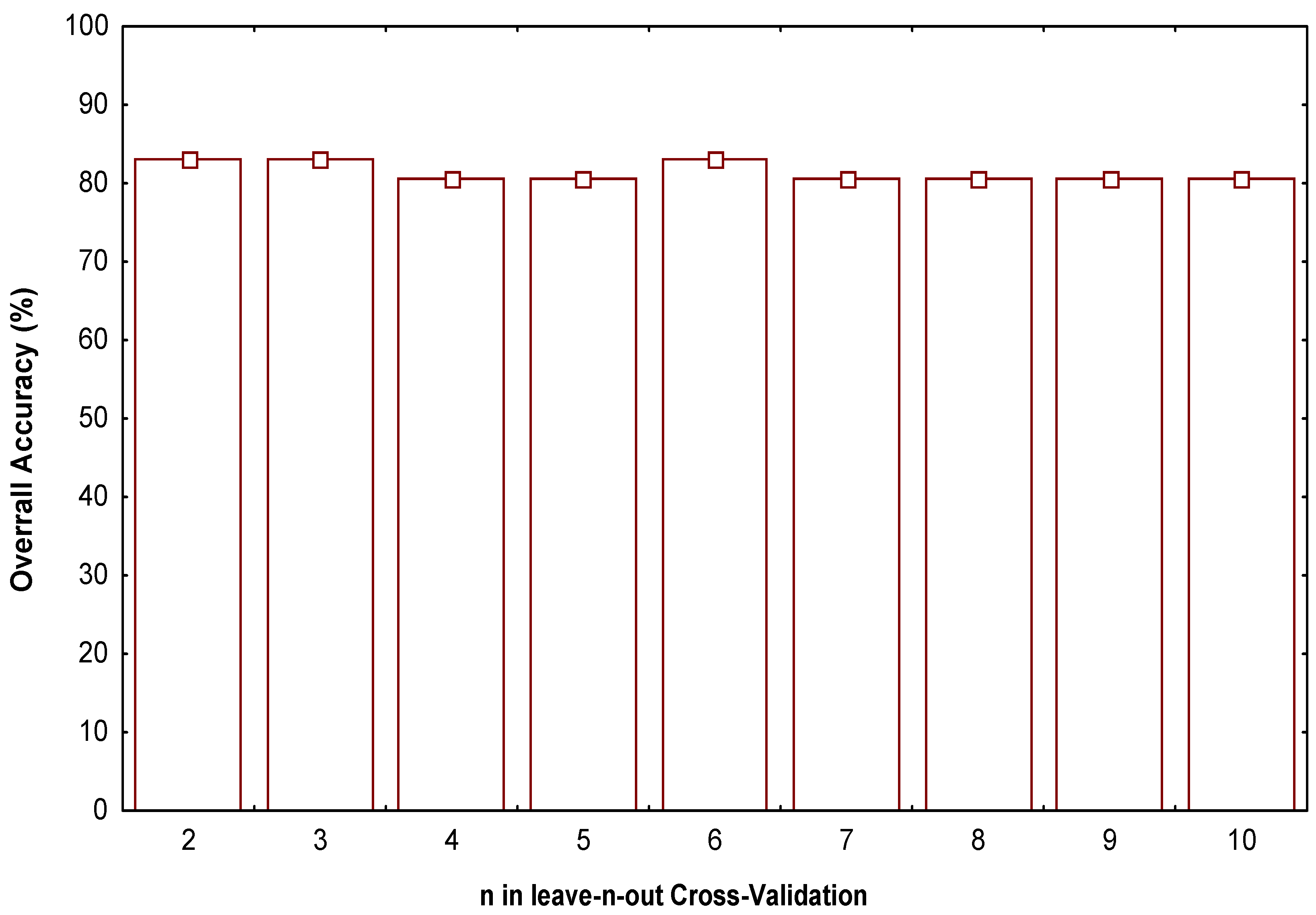
- Test the robustness and predictive power of the QSPR/QSAR equation by using internal and external cross-validation techniques,
- Develop a structural interpretation of the obtained QSAR/QSPR model using macromolecular quadratic indices as molecular descriptors.
Statistical Analysis
Results and Discussion
Classification Model
-0.0170188.Z1q1(xm) +0.0132179.Z2q2(xm)
N = 41 λ = 0.476 D2 = 4.40 F(4,36) = 9.8965 p(F) < 0.0001
-0.008317831.Z1q1(xm) +0.006460173.Z2q2(xm)
N = 41 λ = 0.476 Rcanc = 0.72 χ2 = 27.44 Mean (+) = 0.998 Mean (-) = -1.048

Quantitative Structure-Stability Relationships (QSSP) Study
0.121(±0.048).Z1q1(xm) +8.89x10-5(±3.18x10-5).Z2q10(xm)
-1.369x10-5(±4.11x10-6).Z1q10(xm) +5.998x10-4(±2.157x10-4).Z1q7(xm)
+0.026(±0.014).Z1q2(xm) +3.99x10-5(±3.44x10-5).Z3q8(xm)
N = 41 R = 0.85 R2 = 0.72 s = 5.64 q2 = 0.55 scv = 6.24 F(8.28) = 9.0425 p < 0.0001
-9.4481x10-4.Z1q9(xm) -0.03023.Z3q3(xm) +0.01565.Z3q6(xm)
-0.0037.Z3q8(xm) +0.2131x10-3.Z3q10(xm)
tm (oC)>BKPT = 44.547 +0.0232.Z1q3(xm) -0.0159.Z1q5(xm) +3.046x10-3.Z1q7(xm)
-1.6594x10-4.Z1q9(xm) + 2.5765.Z3q3(xm) +0.0106.Z3q6(xm) -2.3478.Z3q8(xm)
+1.2647x10-4.Z3q10(xm)
N = 41 R = 0.94 R2 = 88.15 Bkpt = 51.32 p < 0.0001
Interpretation of Obtained Models
Conclusions
Acknowledgements
References and Notes
- Fersht, A. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding; W. H. Freeman and Company: New York, 1999. [Google Scholar]
- Sidransky, D.; Hollstein, M. Clinical Implications of the p53 Gene. Ann. Rev. Med. 1996, 47, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Grace, J. B. Bioinformatics: Mathematical Challenges and Ecology. Science 1996, 275, 1861c–1865c. [Google Scholar] [CrossRef]
- Marshall, E. Bioinformatics: Hot Property: Biologists Who Compute. Science 1996, 272, 1730–1732. [Google Scholar]
- Anfinsen, C. B. Principles that Govern the Folding of Protein Chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C. B.; Haber, E.; Sela, M.; White, F. H. The Kinetics of Formation of Native Ribonuclease During Oxidation of the Reduced Polypeptide Chain. Proc. Natl. Acad. Sci. USA 1961, 47, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.–W.; Pan, Q.; Zhang, H.–C.; Wu, Y.–H.; Shi, J.–Y. Support Vector Machines for Predicting Protein Homo–Oligomers by Incorporating Pseudo–Amino Acid Composition. Internet Electron. J. Mol. Des. 2003, 2, 392–402, http://www.biochempress.com. [Google Scholar]
- Zhou, H.; Zhou, Y. Stability Scale and Atomic Solvation Parameters Extracted from 1023 Mutation Experiment. Proteins: Prot. Struc. Funct. Gen. 2002, 49, 483–492. [Google Scholar] [CrossRef]
- Alber, T. Mutational Effects on Protein Stability. Annu. Rev. Biochem. 1989, 58, 765–798. [Google Scholar] [CrossRef] [PubMed]
- Dill, K. A.; Shortle, D. Denatured State of Proteins. Annu. Rev. Biochem. 1991, 60, 795–825. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, D. P. Genetic Studies of Proteins Stability and Mechanisms of Folding. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Matthews, B. W. Structural and Genetic Analysis of Protein Stability. Annu. Rev. Biochem. 1993, 62, 139–160. [Google Scholar] [CrossRef] [PubMed]
- Shortle, D. Denature States of Proteins and Their Roles in Folding and Stability. Curr. Opin. Struct. Biol. 1993, 3, 66–74. [Google Scholar] [CrossRef]
- Knight, K. L.; Bowie, J. U.; Vershon, A. K.; Kelley, R. D.; Sauer, R. T. The Arc and Mnt Repressors: a New Class of Sequence Specific DNA-Binding Protein. J. Biol. Chem. 1989, 264, 3639–3642. [Google Scholar] [PubMed]
- Milla, M. E.; Brown, M. B.; Sauer, R. T. Protein Stability Effects of a Complete Set of Alanine Substitutions in Arc Repressor. Struct. Biol. 1994, 1, 518–523. [Google Scholar] [CrossRef]
- Bowie, J. U.; Sauer, R. T. Equilibrium Dissociation and Unfolding of the Arc Repressor Dimmer. Biochemistry 1989, 28, 7139–7143. [Google Scholar] [CrossRef] [PubMed]
- Milla, M. E.; Saber, R. T. P22 Arc Repressor: Folding Kinetics of a Single Domain, Dimeric Protein. Biochemistry 1994, 33, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Vershon, A. K.; Bowie, J. U.; Karplus, T. M.; Sauer, R. T. Isolation and Analysis of Arc Repressor Mutants: Evidence for an Unusual Mechanism of DNA Binding. Proteins 1986, 1, 302–311. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y.; Romero, V. TOMOCOMD software. Central University of Las Villas. 2002. TOMOCOMD (TOpological MOlecular COMputer Design) for Windows, version 1.0 is a preliminary experimental version; in the future a professional version will be available upon request from Y. Marrero: [email protected]; [email protected].
- Marrero-Ponce, Y. Total and Local Quadratic Indices of the “Molecular Pseudograph`s Atom Adjacency Matrix”: Applications to the Prediction of Physical Properties of Organic Compounds. Molecules 2003, 8, 687–726. [Google Scholar] [CrossRef]
- Marrero-Ponce, Y. Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Definition, Significance-Interpretation and Application to QSAR Analysis of Flavone Derivatives as HIV-1 Integrase Inhibitors. J. Chem. Inf. Comput. Sci. 2004, 44, 2010–2026. [Google Scholar]
- Marrero-Ponce, Y.; Cabrera, M.; Romero, V.; Ofori, E.; Montero, L. A. Total and Local Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”. Application to Prediction of Caco-2 Permeability of Drugs. Int. J. Mol. Sci. 2003, 4, 512–536. [Google Scholar]
- Marrero-Ponce, Y.; Cabrera, M. A.; Romero, V.; González, D. H.; Torrens, F. A New Topological Descriptors Based Model for Predicting Intestinal Epithelial Transport of Drugs in Caco-2 Cell Culture. J. Pharm. Pharm. Sci. 2004, 7, 186–199. [Google Scholar] [PubMed]
- Marrero-Ponce, Y.; Huesca-Guillen, A.; Ibarra-Velarde, F. Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix” and Their Stochastic Forms: A Novel Approach for Virtual Screening and in silico Discovery of New Lead Paramphistomicide Drugs-like Compounds. J. Theor. Chem. (THEOCHEM). [CrossRef]
- Marrero-Ponce, Y. Total and Local (Atom and Atom-Type) Molecular Quadratic Indices: Significance-Interpretation, Comparison to Other Molecular Descriptors and QSPR/QSAR Applications. Bioorg. Med. Chem. 2004, 12, 6351–6369. [Google Scholar] [PubMed]
- Marrero-Ponce, Y.; González-Díaz, H.; Romero-Zaldivar, V.; Torrens, F.; Castro, E. A. 3D-Chiral Quadratic Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix” and their Application to Central Chirality Codification: Classification of ACE Inhibitors and Prediction of σ-Receptor Antagonist Activities. Bioorg. Med. Chem. 2004, 12, 5331–5342. [Google Scholar]
- Marrero-Ponce, Y.; Castillo-Garit, J. A.; Torrens, F.; Romero-Zaldivar, V.; Castro, E. Atom, Atom-Type and Total Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to QSPR/QSAR Studies of Organic Compounds. Molecules. in press.
- Marrero-Ponce, Y.; Castillo-Garit, J.A.; Olazabal, E.; Serrano, H. S.; Morales, A.; Castañedo, N.; Ibarra-Velarde, F.; Huesca-Guillen, A.; Jorge, E.; del Valle, A.; Torrens, F.; Castro, E.A. TOMOCOMD-CARDD, a Novel Approach for Computer-Aided “Rational” Drug Design: I. Theoretical and Experimental Assessment of a Promising Method for Computational Screening and in silico Design of New Anthelmintic Compounds. J. Comput. Aided Mol. Des. Accepted for publication.
- Marrero-Ponce, Y.; Montero-Torres, A.; Romero-Zaldivar, C.; Iyarreta-Veitía, I.; Mayón Peréz, M.; García Sánchez, R. Non-Stochastic and Stochastic Linear Indices of the “Molecular Pseudograph’s Atom Adjacency Matrix”: Application to “in silico” Studies for the Rational Discovery of New Antimalarial Compounds. Bioorg. Med. Chem. [CrossRef]
- Marrero-Ponce, Y.; Castillo-Garit, J.A.; Olazabal, E.; Serrano, H. S.; Morales, A.; Castañedo, N.; Ibarra-Velarde, F.; Huesca-Guillen, A.; Jorge, E.; Sánchez, A. M.; Torrens, F.; Castro, E. A. Atom, Atom-Type and Total Molecular Linear Indices as a Promising Approach for Bioorganic & Medicinal Chemistry: Theoretical and Experimental Assessment of a Novel Method for Virtual Screening and Rational Design of New Lead Anthelmintic. Bioorg. Med. Chem. [CrossRef]
- Marrero-Ponce, Y.; Nodarse, D.; González-Díaz, H.; Ramos de Armas, R.; Romero-Zaldivar, V.; Torrens, F.; Castro, E. Nucleic Acid Quadratic Indices of the “Macromolecular Graph’s Nucleotides Adjacency Matrix”. Modeling of Footprints after the Interaction of Paromomycin with the HIV-1 Ψ-RNA Packaging Region. Int. J. Mol. Sci. 2004, 5, 276–293, (see also CPS: physchem/0401004). [Google Scholar]
- Bowie, J. U.; Sauer, R. T. Identifying Determinants of Folding and Activity for a Protein of Unknown Structure. Proc. Natl. Acad. Sci. USA 1989, 86, 2152–2156. [Google Scholar] [CrossRef] [PubMed]
- Milla, M. E.; Brown, M. B.; Sauer, R. T. P22 Arc Repressor: Enhanced Expression of Unstable Mutants by Addition of Polar C-Terminal Sequences. Protein Sci. 1993, 2, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Bray, D.; Lewis, J.; Raff, M.; Roberts, K.; Watson, J. D. Molecular Biology of the Cell; Garland: New York and London, 1994.
- Freifelder, D. Molecular biology. A Comprehesive Introduction to Prokariotes and Eukaryotes; Editorial Revolucionaria: Havana, 1983. [Google Scholar]
- Lehninger, A. L.; Nelson, D. L.; Cox, M. M. Principles of Biochemistry; Worth Publishers: New York, 1993. [Google Scholar]
- Mathews, C. K.; van Holde, K. E.; Ahern, K. G. Biochemistry; Addison Wesley Longman: San Francisco, 2000. [Google Scholar]
- Stryer, L. W. H. Biochemistry; W. H. Freeman and Company: New York, 1995. [Google Scholar]
- Charton, M.; Charton, B. I. The Dependence of the Chou-Fasman Parameters on Amino Acid Side Chain Structure. J. Theor. Biol. 1983, 102, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, S.; Sjöström, M.; Skagerberg, B.; Wold, S. Peptide Quantitative Structure-Activity Relationship, a Multivariate Approach. J. Med. Chem. 1987, 30, 1126–1135. [Google Scholar] [CrossRef] [PubMed]
- Hellberg, S.; Sjöström, M.; Wold, S. The Prediction of Bradykinin Potentiating Potency of Pentapeptides. An Example of a Peptide Quantitative Structure-Activity Relationship. Acta Chem. Scand., Sect. B 1986, 135–140. [Google Scholar]
- Jonsson, J.; Eriksson, L.; Hellberg, S.; Sjöström, M.; Wold, S. Multivariate Parametrization of 55 Coded and Non-Coded Amino Acid. Quant. Struct. Act. Relat. 1989, 8, 204–209. [Google Scholar] [CrossRef]
- Collantes, E. R.; Dunn III, W. J. Amino Acid Side Chain Descriptors for Quantitative Structure-Activity Relationship Studies of Peptide Analogues. J. Med. Chem. 1995, 38, 2705–2713. [Google Scholar]
- Harary, F. Graph Theory; Addison-Wesley: Reading, MA, 1969; p. 10. [Google Scholar]
- Chartrand, G. Graph as Mathematical Models; Prindle, Weber & Schmidt: Boston, MA, 1977; p. 22. [Google Scholar]
- Wilson, R. J. Introduction to Graph Theory; Oliver & Boyd: Edinburgh, 1972; p. 10. [Google Scholar]
- Trinajstic, N. Chemical Graph Theory, 2nd edition; CRC Press: Boca Raton, FL, 1992; pp. 6–7. [Google Scholar]
- Todeschini, R.; Consonni, V. Handbook of molecular descriptors; Wiley VCH: Weinheim, Germany, 2000. [Google Scholar]
- Randić, M. Generalized Molecular Descriptors. J. Math. Chem. 1991, 7, 155–168. [Google Scholar] [CrossRef]
- STATISTICA version. 5.5; Statsoft, Inc.: Tulsa, OK, USA, 1999.
- McFarland, J. W.; Gans, D. J. Linear Discrminant Analysis and Cluster Significance Analysis. In Comprehesive Medicinal Chemistry; Hansch, C., Sammes, P. G., Taylor, J. B., Eds.; Pergamon Press: Oxford, 1990; pp. 667–689. [Google Scholar]
- Wold, S.; Erikson, L. Statistical Validation of QSAR Results. Validation Tools. In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH Publishers: New York, 1995; pp. 309–318. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graphics. Mod. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Estrada, E.; Patlewicz, G. On the Usefulness of Graph-theoretic Descriptors in Predicting Theoretical Parameters. Phototoxicity of Polycyclic Aromatic Hydrocarbons (PAHs). Croat. Chem. Acta. 2004, 77, 203–211. [Google Scholar]
- van de Waterbeemd, H. Discriminant Analysis for Activity Prediction, In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH Publishers: New York, 1995; pp. 265–282. [Google Scholar]
- Ford, M.-G.; Salt, D.-W. The Use of Canonical Correlation Analysis. In Chemometric Methods in Molecular Design; van de Waterbeemd, H., Ed.; VCH Publishers: New York, 1995; pp. 283–292. [Google Scholar]
- Estrada, E.; Peña, A. In Silico Studies for the Rational Discovery of Anticonvulsant Compounds. Bioorg. Med. Chem. 2000, 8, 2755–2770. [Google Scholar] [CrossRef] [PubMed]
- Estrada, E.; Peña, A.; García-Domenech, R. Designing Sedative/Hynotic Compounds from a Novel Substructural Graph-Theoretical Approach. J. Comput.–Aided Mol. Des. 1998, 12, 583–595. [Google Scholar]
- Estrada, E.; Uriarte, E.; Montero, A.; Teijeira, M.; Santana, L.; De Clercq, E. A. Novel Approach for the Virtual Screening and Rational Design of Anticancer Compounds. J. Med. Chem. 2000, 4, 1975–1985. [Google Scholar]
- González, D. H.; Marrero-Ponce, Y.; Hernández, I.; Bastida, I.; Tenorio, E.; Nasco, O.; Uriarte, E.; Castañedo, N.; Cabrera, M. A.; Aguila, E.; Marrero, O.; Morales, A.; Pérez, M. 3D-MEDNEs: an Alternative "in silico" Technique for Chemical Research in Toxicology. 1. Prediction of Chemically Induced Agranulocytosis. Chem. Res. Toxicol. 2003, 16, 1318–1327. [Google Scholar]
- González, H.; Ramos, R.; Molina, R. Markovian Negentropies in Bioinformatics. 1. A Picture of Footprints after the Interaction of the HIV-1 ψ-RNA Packaging Region with Drugs. Bioinformatics 2003, 16, 2079–2087. [Google Scholar]
- González, H.; Ramos, R.; Molina, R. Vibrational Markovian Modelling of Footprints after the Interaction of Antibiotics with the Packaging Region of HIV Type 1. Bull. Math. Biol. 2003, 65, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Gozalbes, R.; Gálvez, J.; Moreno, A.; Garcia-Domenech, R. Discovery of New Antimalarial Compoundss by Use of Molecular Connectivity Techniques. J. Pharm. Pharmacol. 1999, 51, 111–117. [Google Scholar] [CrossRef] [PubMed]
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Ponce, Y.M.; Marrero, R.M.; Castro, E.A.; Ramos de Armas, R.; Díaz, H. G.; Zaldivar, V.R.; Torrens, F. Protein Quadratic Indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”. 1. Prediction of Arc Repressor Alanine-mutant’s Stability. Molecules 2004, 9, 1124-1147. https://doi.org/10.3390/91201124
Ponce YM, Marrero RM, Castro EA, Ramos de Armas R, Díaz H G, Zaldivar VR, Torrens F. Protein Quadratic Indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”. 1. Prediction of Arc Repressor Alanine-mutant’s Stability. Molecules. 2004; 9(12):1124-1147. https://doi.org/10.3390/91201124
Chicago/Turabian StylePonce, Yovani Marrero, Ricardo Medina Marrero, Eduardo A. Castro, Ronal Ramos de Armas, Humberto González Díaz, Vicente Romero Zaldivar, and Francisco Torrens. 2004. "Protein Quadratic Indices of the “Macromolecular Pseudograph’s α-Carbon Atom Adjacency Matrix”. 1. Prediction of Arc Repressor Alanine-mutant’s Stability" Molecules 9, no. 12: 1124-1147. https://doi.org/10.3390/91201124