Using Variable and Fixed Topological Indices for the Prediction of Reaction Rate Constants of Volatile Unsaturated Hydrocarbons with OH Radicals

Abstract
:Introduction
Experimental
Data set (Unsaturated Hydrocarbons)

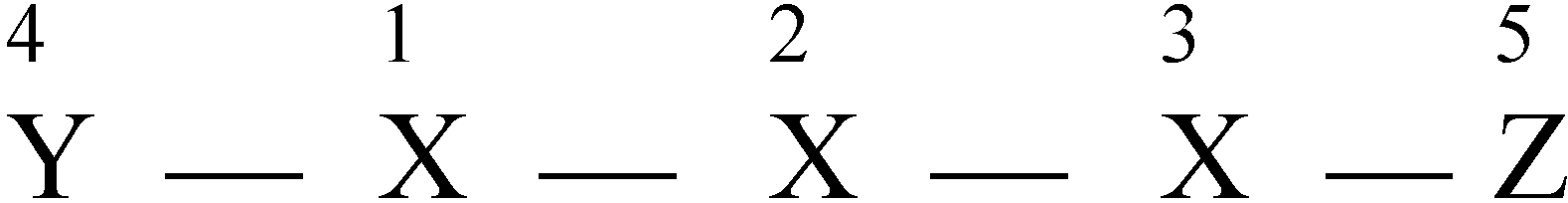{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
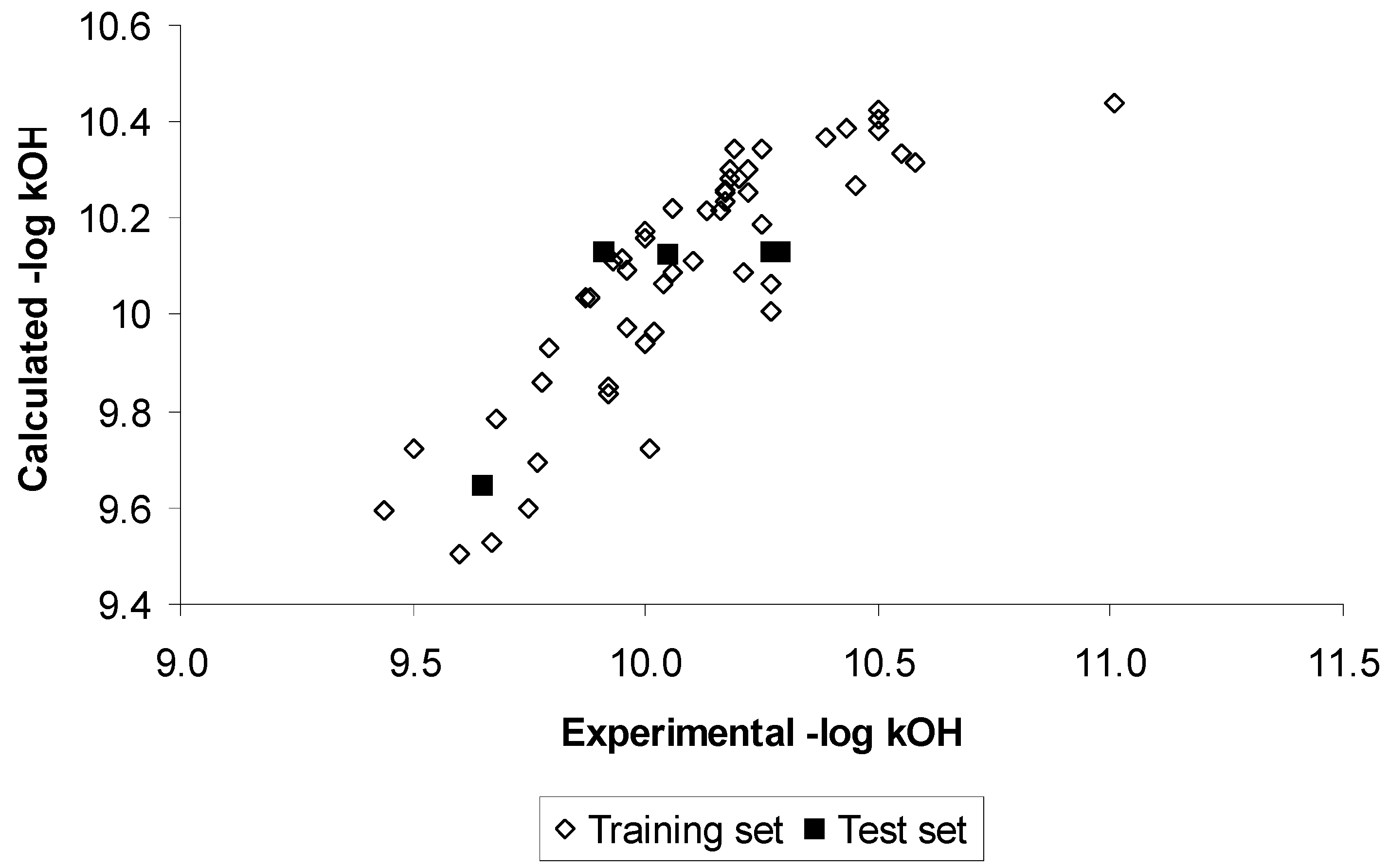{kind=link}
| ID | Name | Experimental | Calculated MLR | Calculated χ1f (model 1) | Calculated χ1f (model 2) |
|---|---|---|---|---|---|
| 1 | α-terpinene | 9.44 | 9.51 | 9.49 | 9.59 |
| 2 | α-phellandrene | 9.50 | 9.73 | 9.57 | 9.72 |
| 3 | trans-β-ocimene | 9.60 | 9.54 | 9.58 | 9.50 |
| 4 | terpinolene* | 9.65 | 9.49 | 9.64 | 9.65 |
| 5 | myrcene | 9.67 | 9.70 | 9.61 | 9.53 |
| 6 | 2,5-dimethyl-2,4-hexadiene | 9.68 | 9.71 | 9.85 | 9.78 |
| 7 | γ-terpinene | 9.75 | 9.55 | 9.57 | 9.60 |
| 8 | Δ-limonene | 9.77 | 9.72 | 9.82 | 9.69 |
| 9 | β-phellandrene | 9.78 | 9.94 | 9.69 | 9.86 |
| 10 | 1,3-cyclohexadiene | 9.79 | 9.88 | 9.86 | 9.93 |
| 11 | trans,trans-2,4-hexadiene | 9.87 | 9.94 | 10.06 | 10.03 |
| 12 | trans –4-methyl-1,3-pentadiene | 9.88 | 10.04 | 10.06 | 10.03 |
| 13 | 2,3-dimethyl-1,3-butadiene* | 9.91 | 9.81 | 10.05 | 10.13 |
| 14 | 2,5-dimethyl-1,5-hexadiene | 9.92 | 9.83 | 9.95 | 9.84 |
| 15 | bicyclo[2.2.1]-2,5-heptadiene | 9.92 | 9.99 | 9.90 | 9.85 |
| 16 | trans –1,3-hexadiene | 9.95 | 10.09 | 10.09 | 10.11 |
| 17 | trans –1,3,5-hexatriene | 9.96 | 9.92 | 9.92 | 9.97 |
| 18 | cis-1,3,5-hexatriene | 9.96 | 9.93 | 9.92 | 9.97 |
| 19 | 2,3-dimethyl-2-butene | 9.96 | 9.96 | 10.18 | 10.09 |
| 20 | 1,3-pentadiene | 10.00 | 10.04 | 10.05 | 10.17 |
| 21 | 2-methyl-1,3-butadiene | 10.00 | 10.10 | 10.16 | 10.16 |
| 22 | 1,4-cyclohexadiene | 10.00 | 9.93 | 9.96 | 9.94 |
| 23 | 1,3,5-cycloheptatriene | 10.01 | 9.56 | 9.70 | 9.72 |
| 24 | 2-methyl-1,5-hexadiene | 10.02 | 10.08 | 10.05 | 9.96 |
| 25 | trans -1,4-hexadiene | 10.04 | 10.11 | 10.12 | 10.06 |
| 26 | 2-methyl-2-pentene* | 10.05 | 10.00 | 10.22 | 10.13 |
| 27 | Δ3-carene | 10.06 | 10.09 | 10.09 | 10.09 |
| 28 | 2-methyl-2-butene | 10.06 | 10.09 | 10.29 | 10.22 |
| 29 | β-pinene | 10.10 | 10.23 | 10.15 | 10.11 |
| 30 | cycloheptene | 10.13 | 10.18 | 10.25 | 10.22 |
| 31 | trans -4-octene | 10.16 | 10.10 | 10.13 | 10.22 |
| 32 | trans -2-heptene | 10.17 | 10.12 | 10.20 | 10.26 |
| 33 | cyclohexene | 10.17 | 10.18 | 10.28 | 10.24 |
| 34 | cyclopentene | 10.17 | 10.26 | 10.31 | 10.26 |
| 35 | trans -2-pentene | 10.18 | 10.14 | 10.33 | 10.30 |
| 36 | 1,3-butadiene | 10.18 | 10.34 | 10.26 | 10.28 |
| 37 | cis -2-pentene | 10.18 | 10.15 | 10.33 | 10.30 |
| 38 | trans -2-butene | 10.19 | 10.28 | 10.39 | 10.34 |
| 39 | 2-methyl-1-pentene | 10.20 | 10.13 | 10.26 | 10.28 |
| 40 | 1,5-hexadiene | 10.21 | 10.36 | 10.15 | 10.09 |
| 41 | 2-methyl-1-butene | 10.22 | 10.13 | 10.32 | 10.30 |
| 42 | trans -4-methyl-2-pentene | 10.22 | 10.13 | 10.26 | 10.26 |
| 43 | 3-methyl-1,2-butadiene | 10.25 | 10.33 | 10.22 | 10.19 |
| 44 | cis -2-butene | 10.25 | 10.29 | 10.39 | 10.34 |
| 45 | α-pinene | 10.27 | 10.02 | 10.01 | 10.01 |
| 46 | 1,4-pentadiene* | 10.27 | 10.35 | 10.22 | 10.13 |
| 47 | camphene | 10.27 | 10.16 | 10.14 | 10.06 |
| 48 | 2-methylpropene* | 10.29 | 10.25 | 10.38 | 10.13 |
| 49 | 1-heptene | 10.39 | 10.38 | 10.23 | 10.37 |
| 50 | 1-hexene | 10.43 | 10.37 | 10.29 | 10.39 |
| 51 | 1,2-pentadiene | 10.45 | 10.42 | 10.25 | 10.27 |
| 52 | 3-methyl-1-butene | 10.50 | 10.40 | 10.35 | 10.38 |
| 53 | 1-pentene | 10.50 | 10.40 | 10.35 | 10.41 |
| 54 | 1-butene | 10.50 | 10.44 | 10.42 | 10.42 |
| 55 | 3,3-dimethyl-1-butene | 10.55 | 10.48 | 10.29 | 10.34 |
| 56 | 1,2-butadiene | 10.58 | 10.57 | 10.31 | 10.31 |
| 57 | propadiene | 11.01 | 10.76 | 10.39 | 10.44 |
| 58 | sabinene | 9.93 | 10.06 | 10.09 | 10.11 |
Calculation and selection of topological indices using CODESSA software
The variable connectivity index (χ1f)

| 1 | 2 | 3 | 4 | 5 | Row sum | |
| 1 | y | 1 | 0 | 1 | 0 | 1+y |
| 2 | 1 | x | 1 | 0 | 0 | 2+x |
| 3 | 0 | 1 | x | 0 | 1 | 2+x |
| 4 | 1 | 0 | 0 | x | 0 | 2+x |
| 5 | 0 | 0 | 1 | 0 | z | 1+z |
 | ||||||
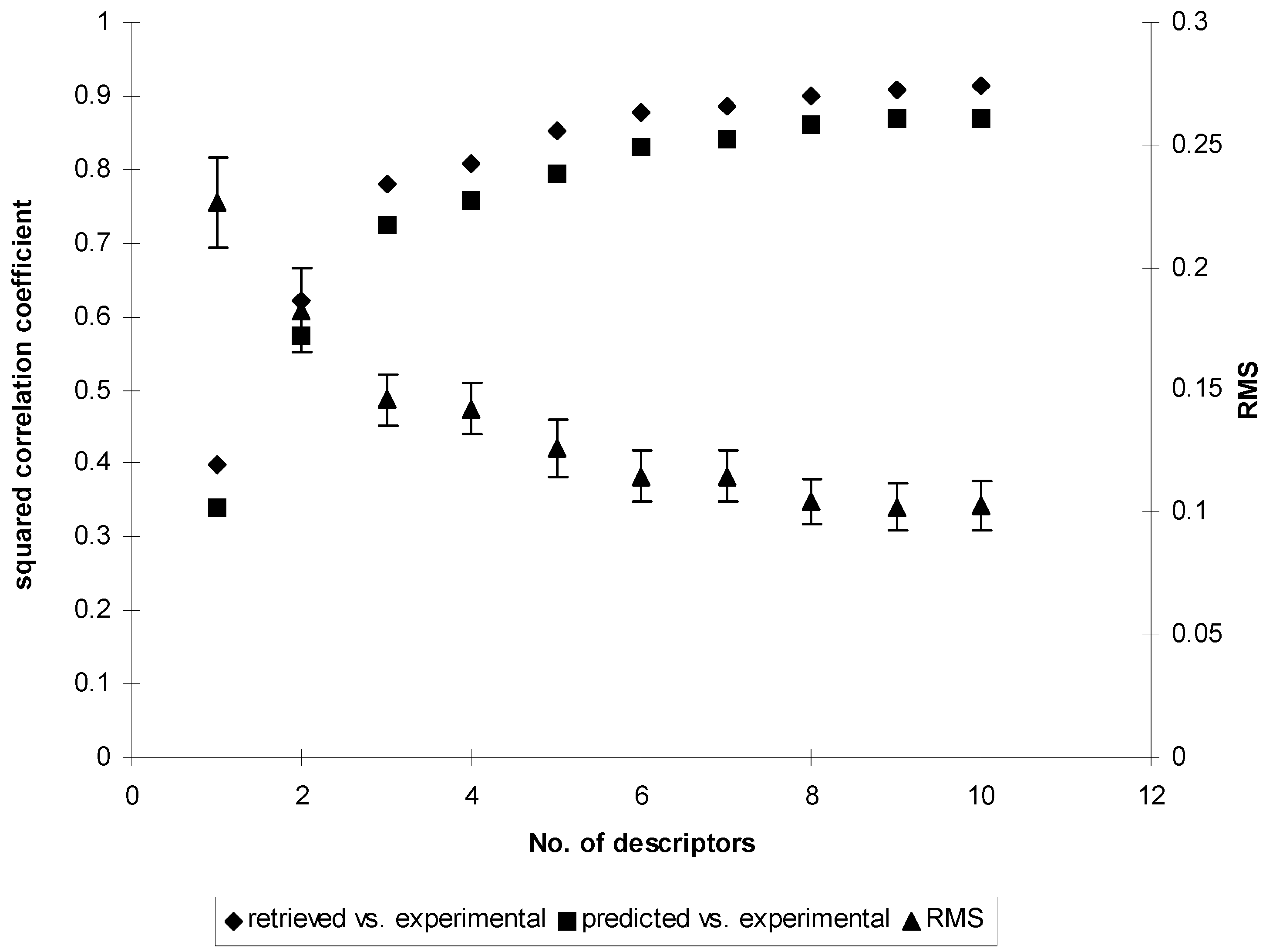
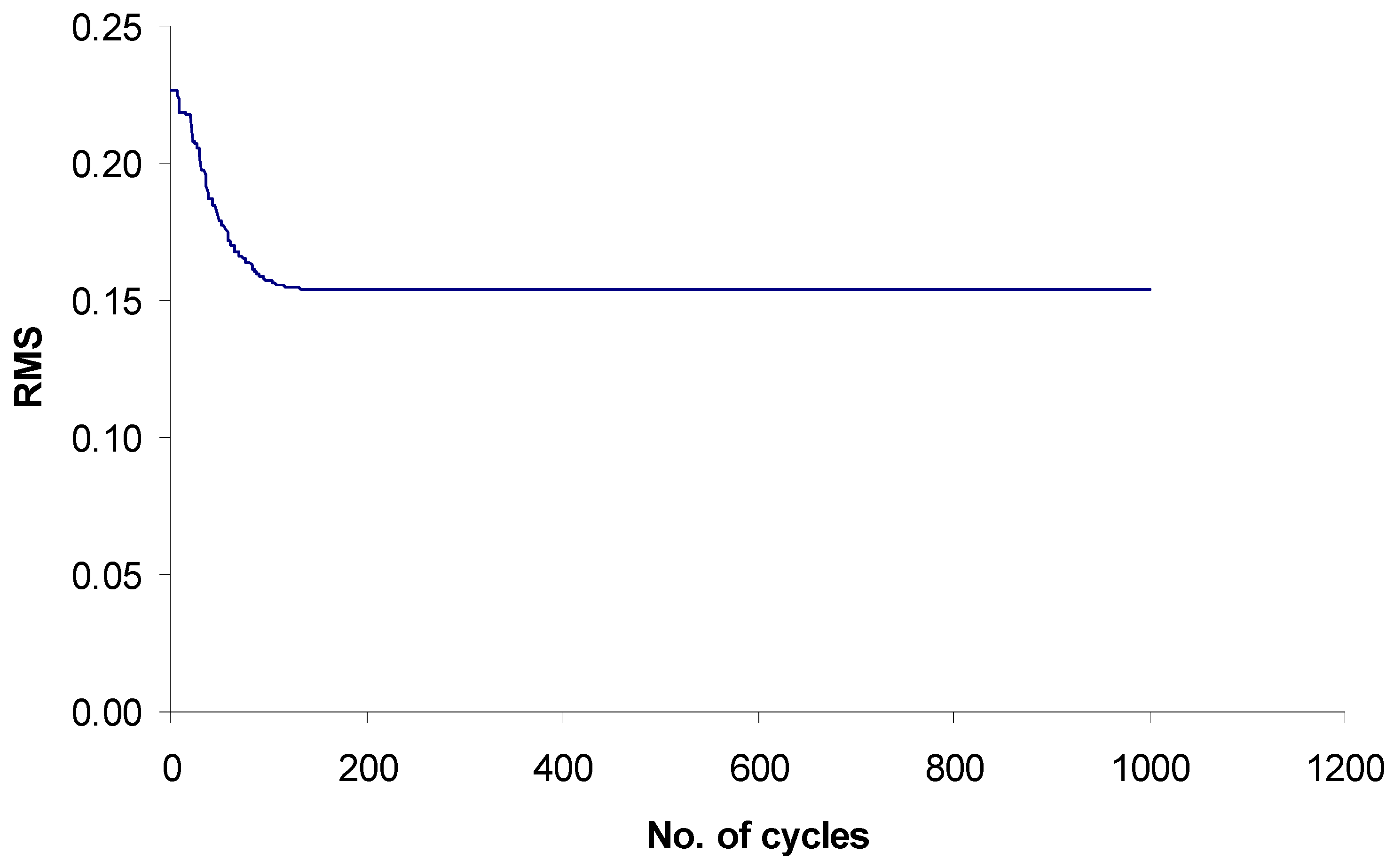
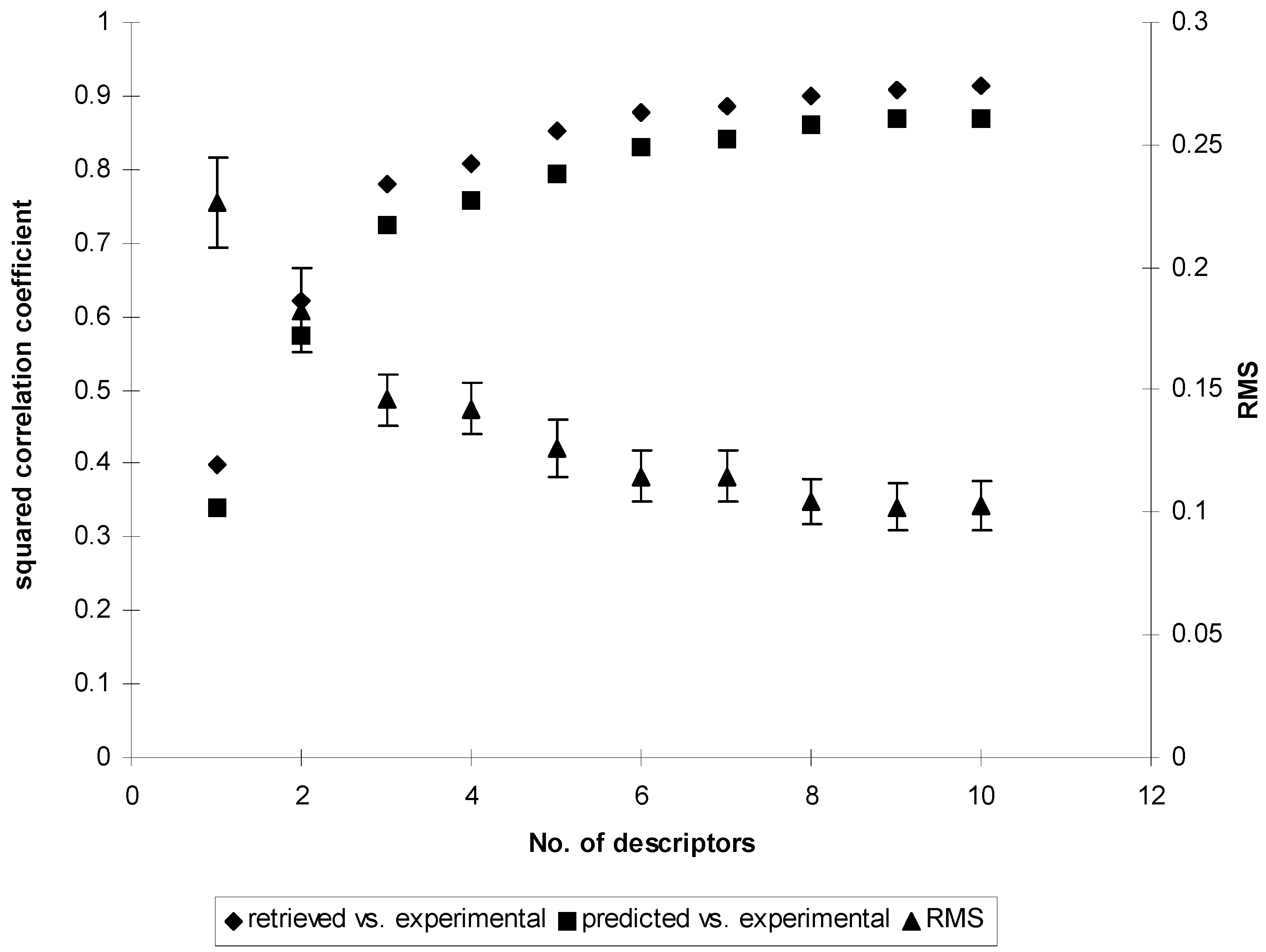
Search for optimal weights in χ1f and evaluation of quality of the models
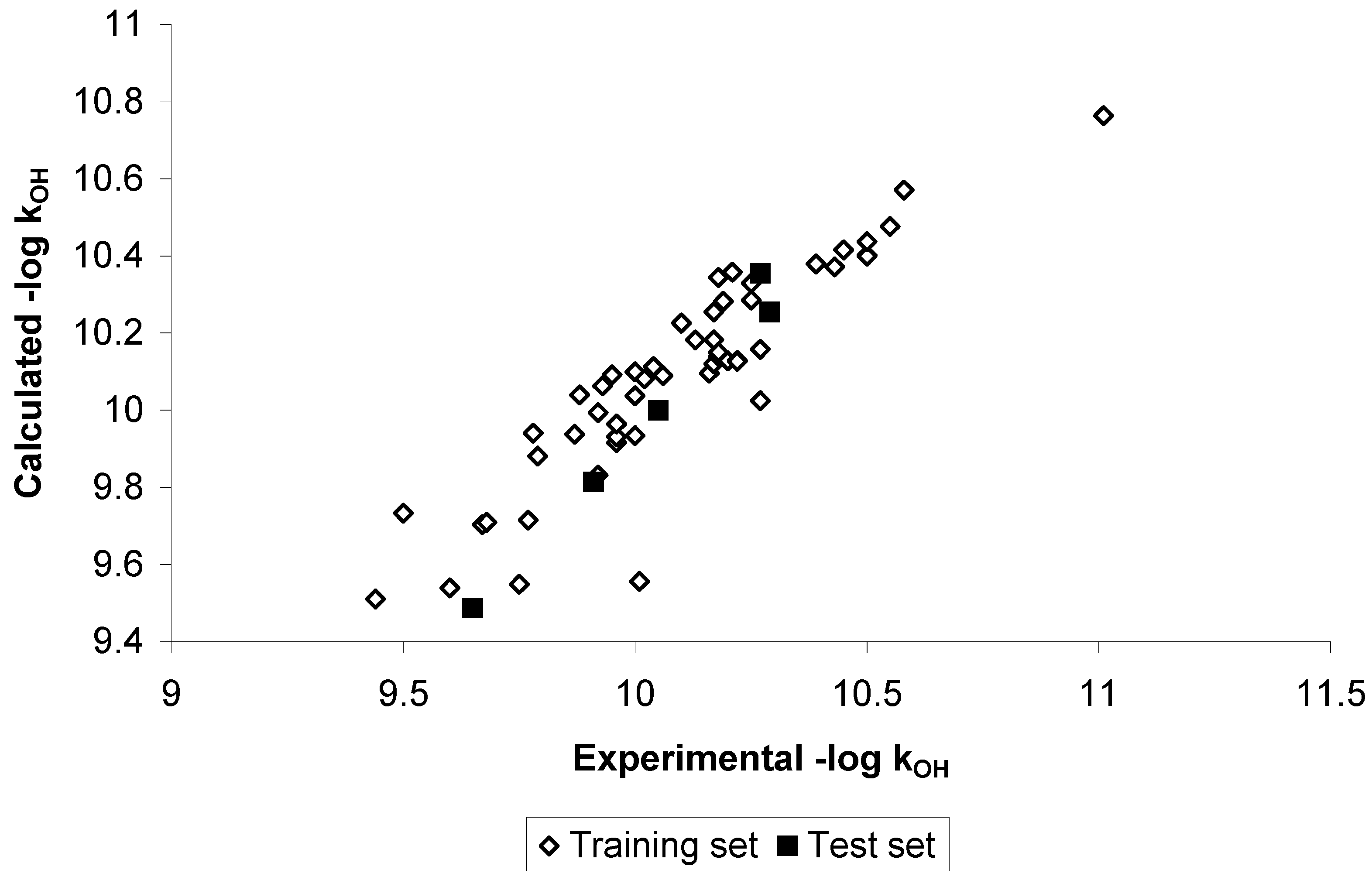
Results and Discussion
| No. | Coefficients | Standard Error | t-test | Partial F | Name of the descriptor |
|---|---|---|---|---|---|
| 0 | 12.23 | 0.179 | 68.37 | 4674 | Intercept |
| 1 | -1.018 | 0.104 | -9.74 | 94.9 | Randic index (order 2) |
| 2 | 1.398 | 0.0988 | 14.14 | 200.1 | Kier & Hall index (order 2) |
| 3 | -0.555 | 0.0993 | -5.59 | 31.2 | Average Complementary Information content (order 0) |
| 4 | 0.405 | 0.0489 | 8.29 | 68.8 | Kier shape index (order 1) |
| 5 | -0.753 | 0.109 | -6.90 | 47.6 | Kier & Hall index (order 0) |
| 6 | 0.000742 | 0.000233 | 3.19 | 10.2 | Gravitation index (all pairs) |
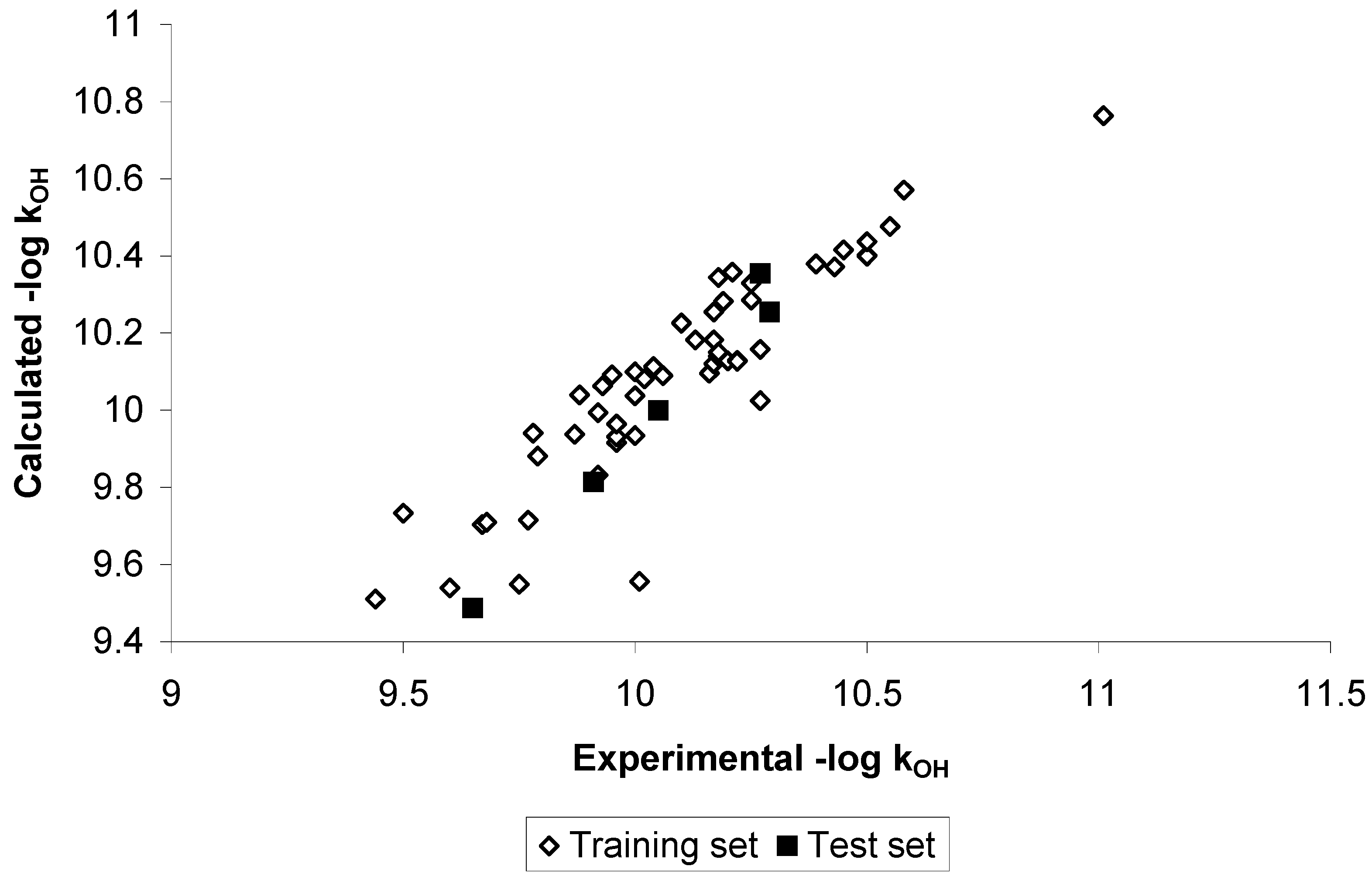
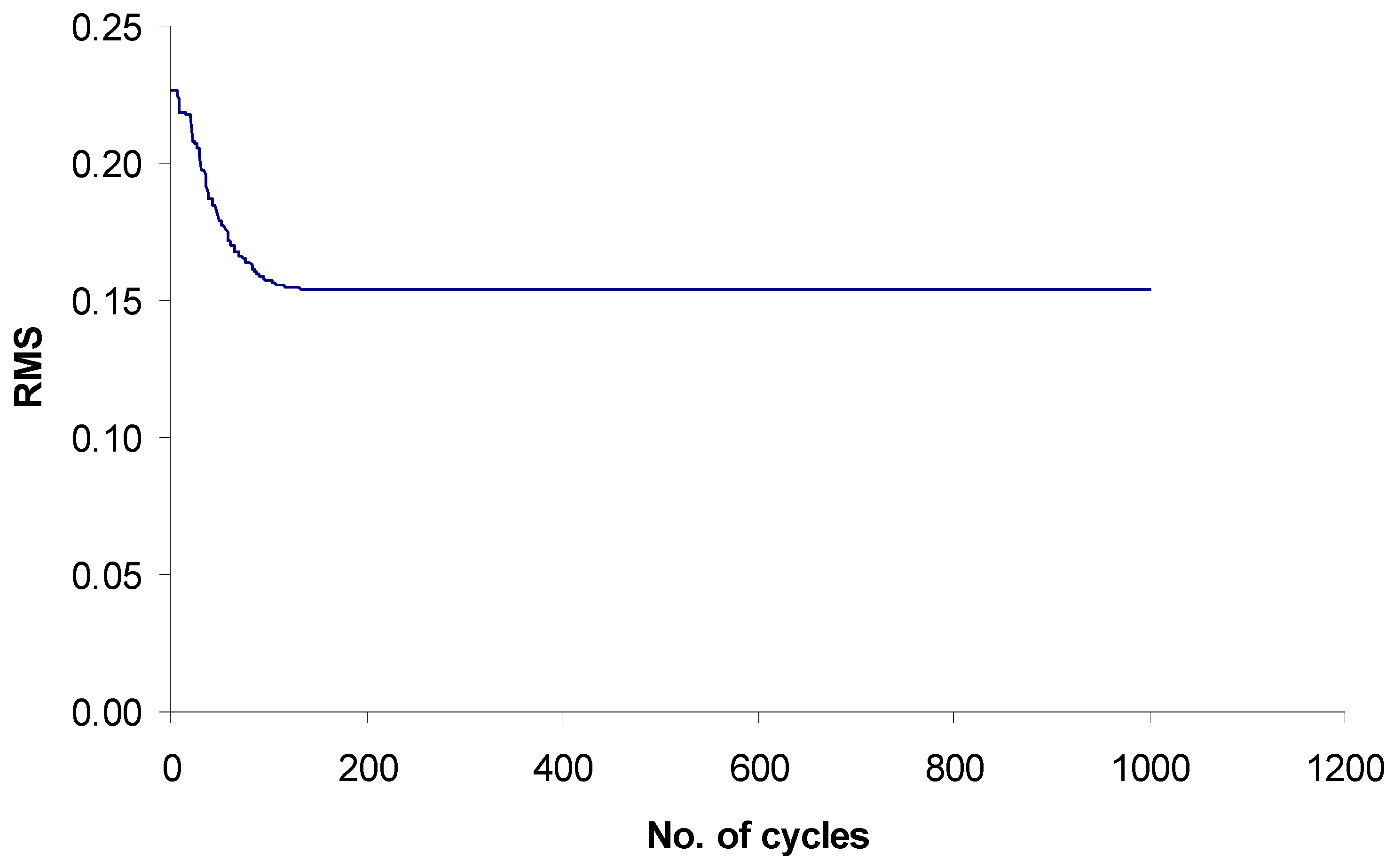

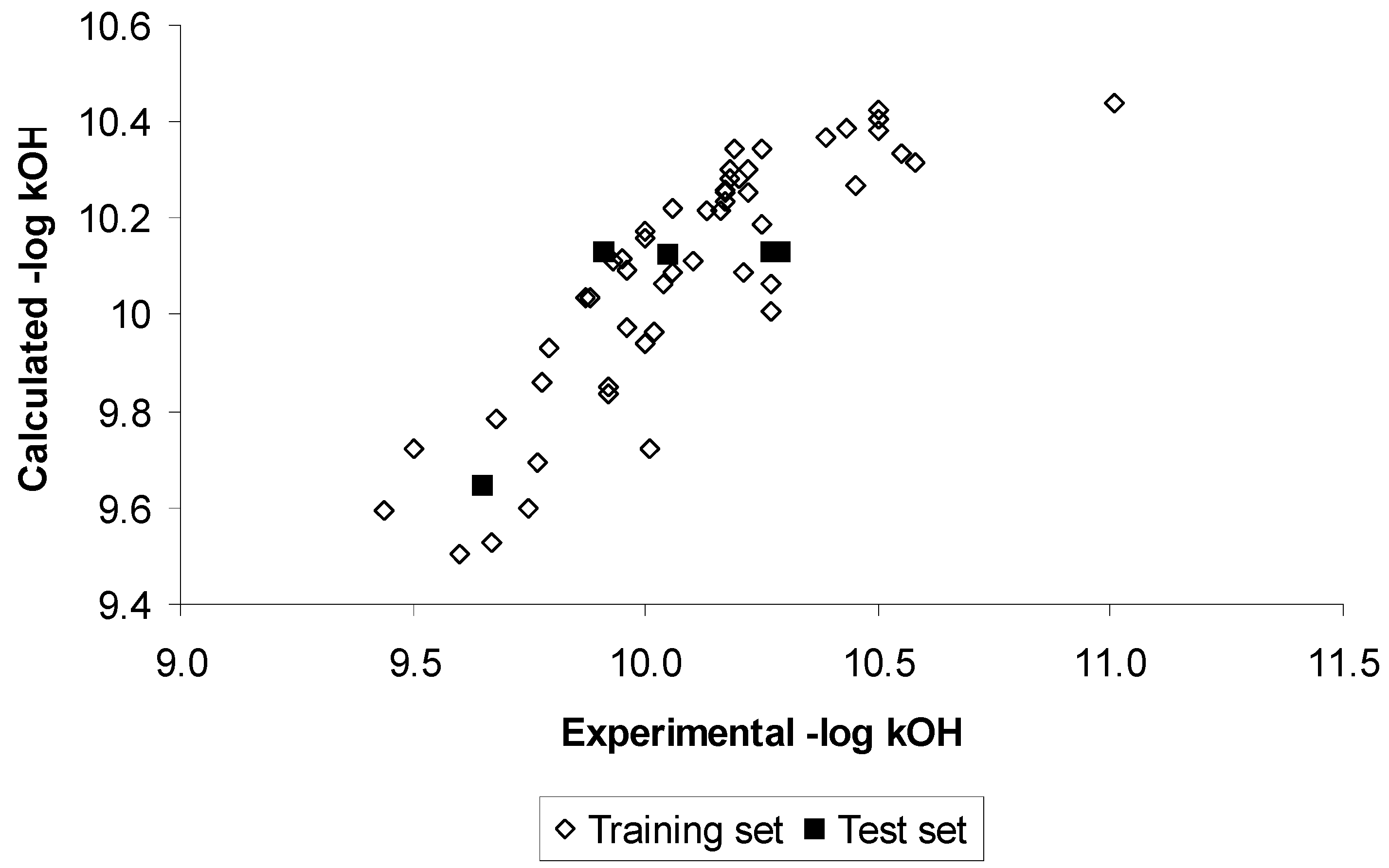
Conclusions
Acknowledgements
References and Notes
- Atkinson, R. Gas-Phase Reactions of the Hydroxyl Radicals. Chem. Rev. 1986, 86, 69–201. [Google Scholar]
- Atkinson, R. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic compounds. J. Phys. Chem. Ref. Data Monograph 1 1989, 1–246. [Google Scholar]
- Atkinson, R. Gas-phase tropospheric chemistry of organic compounds. J. Phys. Chem. Ref. Data Monograph 2 1994, 1–216. [Google Scholar]
- Darnall, K.R.; Atkinson, R.; Pitts, J.N., Jr. Rate Constants for the Reaction of the OH Radical with Selected Alkanes at 300 K. J. Phys. Chem. 1978, 82, 1581–1584. [Google Scholar]
- Atkinson, R. A Structure-activity relationship for the estimation of rate constants for the gas-phase reactions of OH radicals with organic compounds. Int. J. Chem. Kinet. 1987, 19, 799–828. [Google Scholar] [CrossRef]
- Atkinson, R. Estimation of Gas-Phase Hydroxyl Radical Rate Constants for Organic Chemicals. Environ. Toxicol. Chem. 1988, 7, 435–442. [Google Scholar]
- Heickler, J. The correlation of Rate Coefficients for H-Atom Abstraction by OH Radicals with C-H Bond Dissociation Enthalpies. Int. J. Chem. Kinet. 1981, 13, 651–665. [Google Scholar]
- Jolly, G.S.; Paraskevopoulos, G.; Singleton, D.L. Rates of OH radical reactions. XII. The reaction of OH with c-C3H6, c-C5H10, and c-C7H14. Correlation of hydroxyl rate constants with bond dissociation energies. Int. J. Chem. Kinet. 1984, 17, 1–10. [Google Scholar] [CrossRef]
- Cohen, N. The Use of Transition-State Theory to Extrapolate Rate Coefficients for Reaction of OH with Alkanes. Int. J. Chem. Kinet. 1982, 14, 1339–1362. [Google Scholar] [CrossRef]
- Hodson, J. The estimation of the photodegradation of organic compounds by hydroxyl radical reaction rate constants obtained from nuclear magnetic resonance spectroscopy chemical shift data. Chemosphere 1988, 17, 2339–2348. [Google Scholar]
- Grosjean, D.; Williams, E.L. II Environmental persistence of organic compounds estimated from structure-reactivity and linear free-energy relationships-unsaturated aliphatics. Atmos. Environ. 1992, 26A, 1395–1405. [Google Scholar]
- Gaffney, J.S.; Levine, S.Z. Predicting gas-phase organic molecule reaction rates using linear free-energy correlations. I. O(3P) and OH addition and abstraction reactions. Int. J. Chem. Kinet. 1979, 11, 1197–1209. [Google Scholar] [CrossRef]
- Rinke, M.; Wahner, A.; Zetzsch, C.Z. Dependence of the Rate of OH Addition to Aromatics on the Ionization Potential: A Predictive Tool for Rate Constants. J. Photochem. 1981, 17, 142. [Google Scholar] [CrossRef]
- Sekušak, S.; Güsten, H.; Sabljić, A. An ab initio investigation on transition state and reactivity of chloroethane with OH radical. J. Chem. Phys. 1995, 102, 7504–7518. [Google Scholar]
- Sekušak, S.; Güsten, H.; Sabljić, A. An ab initio study on reactivity of chloro ethane with hydroxyl radical: Application of G2 theory. J. Phys. Chem. 1996, 100, 6212–6224. [Google Scholar]
- Klamt, A. Estimation of gas-phase hydroxyl radical rate constants of organic compounds from molecular orbital calculations. Chemosphere 1993, 26, 1273–1289. [Google Scholar]
- Klamt, A. Estimation of gas-phase hydroxyl radical rate constants of oxygenated compounds based on molecular orbital calculations. Chemosphere 1996, 32, 717–726. [Google Scholar]
- Francisco, J.S. Reaction of OH radicals with CH3C(O)H and CF3C(O)H. J. Chem. Soc. Faraday Trans. 1992, 8, 1943–1947. [Google Scholar]
- Melissas, V.S.; Truhlar, D.G. Interpolated variational transition-state theory and semi-classical tunneling calculations of the rate constant of the reaction OH + C2H6 at 200-3000 K. J. Phys. Chem. 1994, 98, 875–886. [Google Scholar]
- King, M.D.; Canosa-Mas, C.E.; Wayne, R.P. Fronier molecular orbital correlations for predicting rate constants between alkenes and the tropospheric oxidants NO3, OH and O3. Phys. Chem. Chem. Phys. 1999, 1, 2231–2238. [Google Scholar]
- King, M.D.; Canosa-Mas, C.E.; Wayne, R.P. A structure-activity relationship (SAR) for predicting rate constants for the reaction of NO3, OH and O3 with monoalkenes and conjugated dienes. Phys. Chem. Chem. Phys. 1999, 1, 2239–2246. [Google Scholar]
- Tosato, M.L.; Chiorboli, C.; Eriksson, L.; Jonsson, L. Multivariate modelling of the rate constant of the gas-phase reaction of haloalkanes with the hydroxyl radical. Sci. Total. Environ. 1991, 109/110, 307–325. [Google Scholar]
- Eriksson, L.; Rännar, S.; Sjöström, M.; Hermens, J.L.M. Multivariate QSARs to model the hydroxyl radical rate constant for halogenated aliphatic hydrocarbons. Environmetrics 1994, 5, 197–208. [Google Scholar]
- Medven, Z.; Güsten, H.; Sabljić, A. Comparative QSAR study on hydroxyl radical reactivity with unsaturated hydrocarbons: PLS versus MLR. J. Chemomet. 1996, 10, 135–147. [Google Scholar]
- Gramatica, P.; Consonni, V.; Todeschini, R. QSAR study on the tropospheric degradation of organic compounds. Chemosphere 1999, 38, 1371–1378. [Google Scholar]
- Bakken, G.A.; Jurs, P.C. Prediction of hydroxyl radical rate constants from molecular structure. J. Chem. Inf. Comput. Sci. 1999, 39, 1064–1075. [Google Scholar] [CrossRef]
- Gramatica, P.; Pilutti, P.; Papa, E. Validated QSAR prediction of OH tropospheric degradation of VOCs: Splitting into training – test set and consensus modeling. J. Chem. Inf. Comput. Sci. 2004, 44, 1794–1802. [Google Scholar] [PubMed]
- Güsten, H. Predicting the abiotic degradability of organic pollutants in the troposphere. Chemosphere 1999, 38, 1361–1370. [Google Scholar]
- Güsten, H.; Medven, Z.; Sekušak, S.; Sabljić, A. Predicting Tropospheric Degradation of Chemicals: From Estimation to Computations. SAR QSAR Environ. Res. 1995, 4, 197–209. [Google Scholar] [PubMed]
- Todeschini, R.; Consonni, V. The Handbook of Molecular Descriptors; in the Series of Methods and Principles in Medicinal Chemistry; Vol. 11; Mannhold, R., Kubinyi, H., Timmerman, H., Eds.; Wiley-VCH: New York, 2000; p. 680. [Google Scholar]
- Katritzky, A.R.; Maran, U.; Lobanov, V.S.; Karelson, M. Structurally diverse quantitative structure-property relationship correlations of technologically relevant physical properties. J. Chem. Inf. Comput. Sci. 2000, 40, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 69, 17. [Google Scholar] [PubMed]
- Platt, J.R. Influence of neighbor bonds on additive bond properties in paraffins. J. Chem. Phys. 1947, 15, 419. [Google Scholar]
- Hosoya, H. Topological Index. A newly Proposed Quantity Characterizing The Topological Nature of structural Isomers of Saturated Hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar]
- Randić, M. Characterisation of molecular branching. J. Am. Chem. Soc. 1975, 97, 6609–6615. [Google Scholar]
- Balaban, A.T. Chemical Applications of Graph Theory; Academic Press: London, 1976; p. 389. [Google Scholar]
- Bonchev, D.; Trinajstić, N. Information-theory, distance matrix, and molecular branching. J. Chem. Phys. 1977, 67, 4517. [Google Scholar]
- Kier, L.B.; Hall, L.H. Molecular Connectivity in Structure-Activity Analysis; Wiley: New York, 1986. [Google Scholar]
- Balaban, A.T.; Ivanciuc, O. Historical development of topological indices, in: Topological Indices and Related Descriptors in QSAR and QSPR; Devillers, J., Balaban, A.T., Eds.; Gordon and Breach: Amsterdam, The Netherlands, 2000; pp. 21–51. [Google Scholar]
- Randić, M. Topological Indices. In The Encyclopedia of Computational Chemistry; Schleyer, P.v.R, Allinger, N.L., Clark, T., Gasteiger, J., Kollman, P.A., Schaefer III, H.F., Schreiner, P.R., Eds.; John Wiley & Sons: Chichester, U.K, 1998; pp. 3018–3032. [Google Scholar]
- Randić, M. On Computation of optimal parameters for multivariate analysis of structure-property relationship. J. Comput. Chem. 1991, 12, 970–980. [Google Scholar]
- Randić, M. Novel graph theoretical approach to heteroatoms in QSAR. Chemometrics & Intel. Lab. Systems 1991, 10, 213–227. [Google Scholar] [CrossRef]
- Randić, M.; Dobrowolski, J.Cz. Optimal molecular connectivity descriptor for nitrogen-containing molecules. Int. J. Quantum Chem. 1998, 70, 1209–1215. [Google Scholar] [CrossRef]
- Randić, M.; Plavšić, D.; Lerš, N. Variable connectivity index for cycle-containing structures. J. Chem. Inf. Comput. Sci. 2001, 41, 657–662. [Google Scholar] [PubMed]
- Pogliani, L. From molecular connectivity indices to semiempirical connectivity terms: Recent trends in graph theoretical descriptors. Chem. Rev. 2000, 100, 3827–3858. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Qunatum-chemical descriptors in QSAR/QSPR studies. Chem. Rev. 1996, 96, 1027–1043. [Google Scholar] [PubMed]
- Katritzky, A.R.; Lobadov, V.S.; Karelson, M.; CODESSA Training Manual, University of Florida, Gainesville, FL. 1995.
- Randić, M. On characterization of chemical structure. J. Chem. Inf. Comput. Sci. 1997, 37, 672–687. [Google Scholar] [CrossRef]
- Klein, D. J. Chemical graph-theoretic cluster expansions. Int. J. Quantum Chem. 1986, S20, 153–183. [Google Scholar] [CrossRef]
- Randić, M. On the representation of molecular graphs by basis graphs. J. Chem. Inf. Comput. Sci. 1992, 32, 57–69. [Google Scholar]
- Keir, L.B.; Wallace, W.J.; Randić, M.; Hall, L.H. Molecular connectivity. V. Connectivity series applied to density. J. Pharm. Sci. 1975, 65, 1226–1230. [Google Scholar]
- Keir, L.B.; Hall, L.H. Molecular connectivity. VII. Specific treatement of heteroatoms. J. Pharm. Sci. 1975, 65, 18066–18090. [Google Scholar]
- See for example: Balaban, A.T. Chemical graphs 48. Topological index J for hetero-atom containing molecules taking into account periodicities of element properties. Math. Chem (MATCH) 1986, 21, 115–122. [Google Scholar]
- Randić, M. The connectivity index 25 years after. J. Mol. Graphics Modelling 2001, 20, 19–35. [Google Scholar]
- Randić, M.; Pompe, M. On characterization of the CC double bond in alkenes. SAR & QSAR in Environ. Res. 1999, 10, 451–471. [Google Scholar] [CrossRef]
- Randić, M.; Basak, S.C. Multiple regression analysis with optimal molecular descriptors. SAR & QSAR in Environ. Res. 2000, 11, 1–23. [Google Scholar] [CrossRef]
- Randić, M. High quality structure-property regressions. Boiling points of smaller alkanes. New J. Chem. 2000, 24, 165–171. [Google Scholar] [CrossRef]
- Randić, M.; Basak, S.C. On construction of high quality structure-property-activity regressions: The boiling points of sulfides. J. Chem. Inf. Comput. Sci. 2000, 40, 899–905. [Google Scholar] [PubMed]
- Randić, M. On the variable connectivity index and other variable molecular descriptors. In 220th ACS Meeting, Washington D. C., August 20-243, 2000.
- Randić, M.; Mills, D.; Basak, S.C. On use of variable connectivity index for characterization of amino acids. Int. J. Quantum Chem. 2000, 80, 1199–1209. [Google Scholar] [CrossRef]
- Randić, M.; Pompe, M. On variable molecular descriptors based on distance related matrices. J. Chem. Inf. Comput. Sci. 2001, 41, 575–581. [Google Scholar] [PubMed]
- Randić, M.; Basak, S.C. On use of the variable connectivity index 1χf in QSAR: Toxicity of aliphatic ethers. J. Chem. Inf. Comput. Sci. 2001, 41, 614–618. [Google Scholar] [PubMed]
- Randić, M.; Pompe, M. The variable connectivity index 1χf versus the traditional molecular descriptors: A comparative study of 1χf against descriptors of CODESSA. J. Chem. Inf. Comput. Sci. 2001, 41, 631–638. [Google Scholar] [PubMed]
- Randić, M.; Plavšić, D.; Lerš, N. Variable connectivity index for cycle-containing structures. J. Chem. Inf. Comput. Sci. 2001, 41, 657–662. [Google Scholar] [PubMed]
- Massart, D.L.; Vandeginste, D.G.M.; Deming, S.M.; Michotte, Y.; Kaufman, L. Chemometrics: A textbook, Data handling in science and technology; Vol 2; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Pompe, M.; Veber, M.; Randić, M.; Balaban, A.T. Using Variable and Fixed Topological Indices for the Prediction of Reaction Rate Constants of Volatile Unsaturated Hydrocarbons with OH Radicals. Molecules 2004, 9, 1160-1176. https://doi.org/10.3390/91201160
Pompe M, Veber M, Randić M, Balaban AT. Using Variable and Fixed Topological Indices for the Prediction of Reaction Rate Constants of Volatile Unsaturated Hydrocarbons with OH Radicals. Molecules. 2004; 9(12):1160-1176. https://doi.org/10.3390/91201160
Chicago/Turabian StylePompe, Matevž, Marjan Veber, Milan Randić, and Alexandru T. Balaban. 2004. "Using Variable and Fixed Topological Indices for the Prediction of Reaction Rate Constants of Volatile Unsaturated Hydrocarbons with OH Radicals" Molecules 9, no. 12: 1160-1176. https://doi.org/10.3390/91201160