New Imide 5-HT1A Receptor Ligands – Modification of Terminal Fragment Geometry

Department of Medicinal Chemistry, Institute of Pharmacology of the Polish Academy of Sciences, 12 Smętna Street, 31-343 Kraków, Poland
*
Author to whom correspondence should be addressed.
Molecules 2004, 9(3), 170-177; https://doi.org/10.3390/90300170
Submission received: 14 January 2004
/
Accepted: 19 January 2004
/
Published: 28 February 2004
(This article belongs to the Special Issue Biologically Relevant Heterocyclic Compounds)
Abstract
:Two sets of new o-methoxyphenylpiperazine (MPP; series a) and 1,2,3,4-tetrahydroisoquinoline (THIQ; series b) derivatives, containing various imide moieties derived from NAN190, were synthesized and evaluated in vitro for their ability to bind to the serotonin 5-HT1A and 5-HT2A receptors. All new derivatives from series a demonstrated high 5-HT1A affinities, whereas THIQ analogues were much less active. With respect to 5‑HT2A receptors, three MPP derivatives presented moderate activity but the rest of the investigated compounds were practically inactive. The influence of changes in terminus geometry on 5-HT1A receptor affinity was analyzed in regard to model compounds NAN190 and MM199.
Introduction
During the last decade a large number of structurally different compounds have been proposed as 5‑HT1A receptor ligands. Among these, 4-substituted long chain 1-arylpiperazines (LCAPs) have been extensively studied. In particular, much effort has been devoted to the role of the terminal part in a ligand-receptor interaction, and in consequence, a great many different fragments were used [1]. The imide containing fragments constituted one of the most thoroughly investigated termini type and among others, compounds like buspirone (anxiolytic drug; 5-HT1A partial agonist [2]) or NAN190 (postsynaptic antagonist [3,4]) were found (Figure 1).
Figure 1.
Structure and binding constants (Ki) of the selected 5-HT1A receptor ligands.
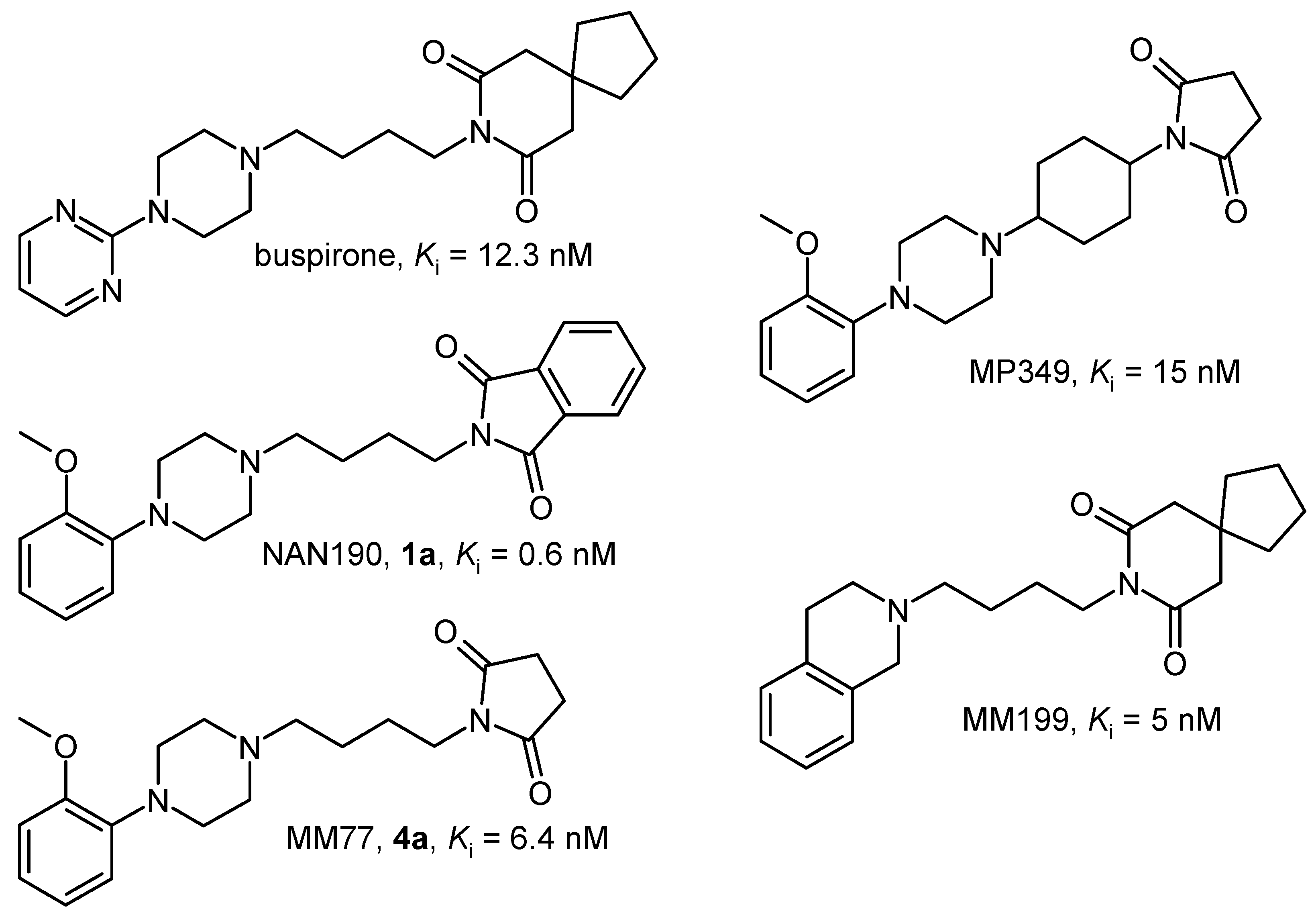
Our systematic SAR studies of the long chain 5-HT1A ligands resulted in identification of several new imide derivatives with interesting biological activity. Two NAN190 analogues: MM77 (postsynaptic antagonist [5]) and its constrained form MP349 (full antagonist [6,7]) exhibited anxiolytic-like activity in some animal models [7,8]. Replacement of 1-pyrimidinylpiperazine fragment of buspirone with 1,2,3,4-tetrahydroisoquinoline (THIQ) led to the compound MM199 [9] (Figure 1), for which anxiolytic-like and antidepressant-like effects in rats were described [10]. Continuing exploration of this type of agents we synthesized two sets of new o-methoxyphenylpiperazine (MPP) and THIQ derivatives and evaluated their affinity for 5-HT1A and 5-HT2A receptors.
Results and Discussion
The structures of NAN190 and its simplified analog MM77 were chosen as lead molecules for modification of imide terminus. All the new compounds were examined in vitro for their ability to displace [3H]-OH-DPAT and [3H]-ketanserine binding to rat 5-HT1A and 5-HT2A receptors respectively, as described in the Experimental Section. The results are presented in Table 1, where additionally, the affinities of parent compounds and previously reported derivatives 1b, 2a, 2b and 4b are also included [9,11].

{kind=link}
{kind=link}
| No. | Imide moiety | a: |  | b: |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ki ± SEM (nM) | Ki ± SEM (nM) | |||||||||||||
| 5-HT1A | 5-HT2A | 5-HT1A | 5-HT2A | |||||||||||
| 1 |  | 0.55±0.14a,b | 451±65 | 355±28b | 2770±130 | |||||||||
| 2 |  | 4.0±0.2b | 109±22 | 50±11b | 880±12 | |||||||||
| 3 |  | 1.7±0.5 | 185±42 | 157±17 | 1780±25 | |||||||||
| 4 |  | 6.4±0.3c | 1510±95 | 2920±90d | NT | |||||||||
| 5 |  | 6.8±0.5 | 690±40 | 453±15 | 4000±30 | |||||||||
| 6 |  | 7.5±0.7 | 835±45 | 690±20 | 4400±40 | |||||||||
| 7 |  | 46±3 | 86±2 | 620±18 | 1230±34 | |||||||||
a Ki value according to Glennon et al. was 0.58 nM [3]. bKi value from ref 11. c ref 5. d ref 9.
All four newly synthesized MPP derivatives (3a and 5a–7a) showed a high affinity for 5-HT1A receptors (Ki = 1.7–46 nM), however, lower than that of NAN190. This is thus another confirmation that the flat phthalimide terminus optimally binds to the respective receptor region. All modifications of this fragment, namely saturation (2a and 3a), removal of the aromatic ring (4a), changes in its position (5a and 6a) or enlargement (7a) decreased the observed 5-HT1A receptor affinity. Compound 7a, the least active at the 5-HT1A sites, was found to be the most potent 5-HT2A ligand. All the other MPP derivatives displayed a moderate to low affinity for 5-HT2A receptors (Ki = 109–1510 nM), and except for 2a, were at the same time at least 100-fold selective 5-HT1A agents. This is not unexpected, since it is known that the MPP system prefers 5-HT1A binding sites.
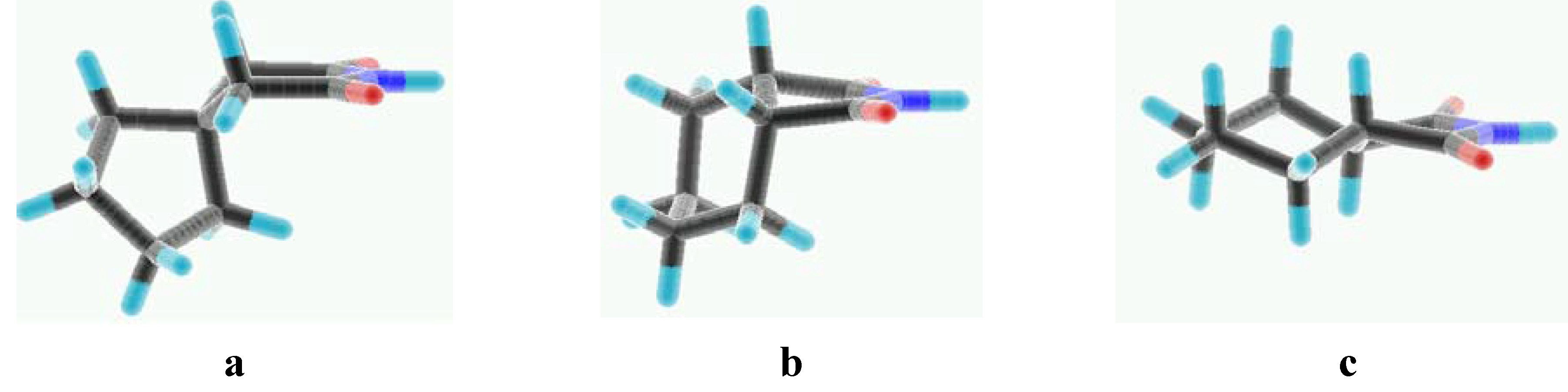
The THIQ moiety has often been used as a replacement for the arylpiperazine fragment in our studies on the role of individual pharmacophoric groups of LCAPs. Although THIQ itself does not show any affinity for the 5-HT1A receptors (Ki > 50 000 nM) [12] its substitution with butyl-azaspiro[4.5]decane-7,9-dione (MM199, Figure 1) increased the affinity to nanomolar level [9]. The same was observed in the case of the 1-adamantoyloaminobutyl THIQ derivative, for which Ki = 0.95 nM was determined [13]. It was then concluded that bulky alicyclic termini should be especially well accommodated by the 5-HT1A binding site. This finding is additionally confirmed by the results obtained for the second group of imide derivatives b. Indeed, the highest 5-HT1A receptor affinity was observed for the alicyclic systems (2b and 3b), whereas aromatic groups were less favorable, although they were better than the small succinimide alone (4b, Ki = 2920 nM). Thus, the replacement of MPP by a THIQ fragment caused a decrease of 5-HT1A affinity and we did not obtain any more active compounds than the previously examined 2b [11]. A more detailed comparison of terminal fragments of MM199, 2b and 3b reveals that their geometry may play a role, and that a bent configuration of the cycloalkyl part (Figure 2) is preferred. With regard to 5-HT2A receptors, we did not detect a significant activity in the investigated THIQ derivatives.
Figure 2.
The geometry of terminal fragments: azaspiro[4.5]decane-7,9-dione (a), cis- (b) and trans-1,2-cyclohexanedicarboxyimide (c).
Figure 2.
The geometry of terminal fragments: azaspiro[4.5]decane-7,9-dione (a), cis- (b) and trans-1,2-cyclohexanedicarboxyimide (c).
As a result of present investigation three new highly active 5-HT1A ligands were found (3a, 5a and 6a), which displayed a 100-fold selectivity over 5-HT2A receptors. During the preparation of this manuscript, Hackling and coworkers published a paper in which compound 7a was investigated as a dopamine D2 and D3 receptor ligand [14]. In our hands this derivative was a dual 5-HT1A/5-HT2A ligand and based on the data obtained by Hackling, it is also a potent but unselective D2/D3 agent (Ki = 40 and 29 nM, respectively). Pharmacological work is currently in progress to evaluate the 5-HT1A functional profile of these four compounds.
Experimental
General
Melting points were determined on a Boetius apparatus and are uncorrected. 1H-NMR spectra were obtained on a Varian EM-360L (60 MHz) in the CDCl3 solution with Me4Si as an internal reference. Elemental analyses were performed in the Institute of Organic Chemistry PAS (Warsaw, Poland), and were within 0.5% of the theoretical values. The syntheses of 2-(4-aminobutyl)-1,2,3,4-tetrahydroisoquinoline [9] and 4-(4-aminobutyl)-1-(2-methoxyphenyl)piperazine [15] have been previously reported.
General method for the preparation of compounds 3a, 3b, 5a, 5b, 6a, 6b, 7a and 7b.
Equimolar amounts (1 mmol) of the respective, commercially available acid anhydride and 4-(4-aminobutyl)-1-(2-methoxyphenyl)piperazine or 2-(4-aminobutyl)-1,2,3,4-tetrahydroisoquinoline were refluxed in xylene (20 mL) for 3 h. After the mixture was allowed to cool to room temperature, the xylene was evaporated under reduced pressure and the products were isolated on a silica gel column. Free bases were converted into the hydrochloride salts in CHCl3 or acetone solution by treatment with excess of Et2O saturated with gaseous HCl.
4-{4-[2-(trans-1,2-Cyclohexanedicarboxyimido)]butyl}-1-(2-methoxyphenyl)piperazine (3a).
Oil, yield (base) 90%; M.p. (salt) 192-194°C (acetone); Rf = 0.45 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.1-6.7 (m, 4H, arom), 3.9 (s, 3H, OCH3), 3.7-3.3 (m, 2H), 3,3-2.9 (m, 4H), 2.9-2.1 (m, 8H), 2.1-1.2 (m, 12H); C23H33N3O3·2HCl (472.03), Calcd. C 58.52, H 7.90, N 8.90; Found C 58.37, H 7.52, N 8.90.
1-(2-Methoxyphenyl)-4-[4-(3-phenylsuccinimido)butyl]piperazine (5a).
Oil, yield (base) 76%; M.p. (salt) 205-207°C (acetone); Rf = 0.46 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.5-7.1 (m, 5H, arom), 7.1-6.7 (m, 4H, arom), 4.2-3.8 (m, 1H), 3.8 (s, 3H, OCH3), 3.8-3.3 (m, 2H), 3,3-2.2 (m, 12H), 2.9-1.3 (m, 4H); C25H31N3O3·2HCl (494.464), Calcd. C 60.72, H 6.72, N 8.49; Found C 60.24, H 6.90, N 8.43.
1-(2-Methoxyphenyl)-4-[4-(3-phenylmaleicimido)butyl]piperazine (6a).
Oil, yield (base) 56%; M.p. (salt) 209-210°C (acetone); Rf = 0.53 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.5-7.0 (m, 5H, arom), 7.0-6.8 (m, 4H, arom), 4.1-3.6 (m, 3H), 3.8 (s, 3H, OCH3), 3,3-2.9 (m, 4H), 2.9-2.1 (m, 10H), 1.9-1.3 (m, 4H); C26H33N3O3·2HCl (508.49), Calcd. C 61.41, H 6.93, N 8.26; Found C 61.00, H 6.99, N 8.08.
1-(2-Methoxyphenyl)-4-[4-(1,8-naphthalimido)butyl)]piperazine (7a).
Oil, yield (base) 84%; M.p. (salt) 280-281°C (acetone); Rf = 0.52 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 8.6(d, J=8Hz, 2H, arom), 8.2 (d, J=8Hz, 2H, arom), 7.7 (t, J=8Hz, arom), 7.1-6.7 (m, 4H, arom.), 4.5-4.0 (m, 2H), 3.9 (s, 3H, OCH3), 3,3-2.9 (m, 4H), 2.9-2.3 (m, 6H), 2.1-1.5 (m, 4H); C27H32 N3O3·2HCl (552.926), Calcd. C 58.65, H 5.83, N 7.60; Found C 58.83, H 6.36, N 7.63.
2-{4-[2-(trans-1,2-Cyclohexanedicarboxyimido)]butyl}-1,2,3,4-tetrahydroisoquinoline (3b).
Oil, yield (base) 79%; M.p. (salt) 168-170°C (acetone); Rf = 0.49 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.3-6.9 (m, 4H, arom), 3.7-3.3 (m, 4H) 3.1-2.0 (m, 8H), 2.0-1.2 (m, 12H); C21H28N2O2·HCl·0.5H2O (385.919), Calcd. C 65.35, H 7.83, N 7.26; Found C 64.99, H 7.89, N 7.08.
2-[4-(3-Phenylsuccinimido)butyl]-1,2,3,4-tetrahydroisoquinoline (5b).
Oil, yield (base) 81%; M.p. (salt) 127-129°C (acetone); Rf = 0.39 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.6-7.2 (m, 5H, arom), 7.2-7.0 (m, 4H, arom), 4.1-3.3 (m, 5H), 3.3-2.3 (m, 8H), 2.1-1.3 (m, 4H); C23H26N2O2 · HCl · 0.25H2O (403.439), Calcd. C 68.47, H 6.87, N 6.94; Found C 68.47, H 6.89, N 6.85.
2-[4-(3-phenylmaleicimido)butyl]-1,2,3,4-tetrahydroisoquinoline (6b).
Oil, yield (base) 47%; M.p. (salt) 121-123°C (acetone); Rf = 0.43 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 7.6-7.2 (m, 5H, arom), 7.2-6.9 (m, 4H, arom), 4.2-3.5 (m, 5H), 3.3-2.0 (m, 10H), 1.9-1.4 (m, 4H); C24H28N2O2·HCl·1.25H2O (435.482), Calcd. C 66.19, H 7.29, N 6.43; Found C 66.43, H 7.26, N 6.16.
2-[4-(1,8-naphthalimido)butyl]-1,2,3,4-tetrahydroisoquinoline (7b).
Oil, yield (base) 40%; M.p. (salt) 265-267°C (acetone); Rf = 0.43 (CHCl3/MeOH 19:1); 1H-NMR (base): δ (ppm) = 8.6 (d, J=8Hz, 2H, arom), 8.2 (d, J=8Hz, 2H, arom), 7.7 (t, J=8Hz, 2H arom), 7.3-6.9 (m, 4H, arom), 4.5-4.0 (m, 2H, CH2), 3.8-3.5 (m, 2H), 3,1-2.4 (m, 6H), 2.1-1.5 (m, 4H); C25H24 N2O2· HCl·0.5H2O (429.942), Calcd. C 69.84, H 6.09, N 6.51; Found C 70.17, H 5.63, N 6.17.
Radioligand binding studies
The in vitro affinity of the investigated compounds for 5-HT1A and 5-HT2A receptors was assessed on the basis of their ability to displace [3H]-8-OH-DPAT (222 Ci/mmol, Amersham, England) and [3H]-ketanserin (66.4 Ci/mmol, NEN Chemicals, USA), respectively. Radioligand binding experiments were carried out on rat brain using tissues from the hippocampus for 5-HT1A receptors, and from the cortex for 5-HT2A receptors, according to the previously published procedures [16].
Ki values were determined from at least three competitive binding experiments in which 10–14 sample concentrations, run in triplicate, were used. The Cheng and Prusoff [17] equation was used for Ki calculations.
References
- Lopez-Rodriguez, M.L.; Ayala, D.; Benhamu, B.; Morcillo, M.J.; Viso, A. Arylpiperazine Derivatives Acting at 5-HT1A Receptors. Curr. Med. Chem. 2002, 9, 443. [Google Scholar]
- Goa, K.L.; Ward, A. Buspirone a preliminary review of its pharmacological properties and therapeutic efficacy as an anxiolytic. Drugs 1986, 32, 114. [Google Scholar]
- Glennon, R.A.; Naiman, N.A.; Pierson, M.E.; Titeler, M.; Lyon, R.A.; Weisberg, E. NAN-190: an arylpiperazine analog that antagonizes the stimulus effects of the 5-HT1A agonist 8-hydroxy-2-(di-n- propylamino)tetralin (8-OH- DPAT). Eur. J. Pharmacol. 1988, 154, 339. [Google Scholar]
- Zifa, E.; Fillion, G. 5-Hydroxytryptamine receptors. Pharmacol. Rev. 1992, 44, 401. [Google Scholar]
- Mokrosz, M.J.; Chojnacka-Wójcik, E.; Tatarczyńska, E.; Kłodzińska, A.; Filip, M.; Boksa, J.; Charakchieva-Minol, S.; Mokrosz, J.L. 1-(2-Methoxyphenyl)-4-[(4-succinimido)butyl]piperazine (MM-77): A New, Potent, Postsynaptic Antagonist of 5-HT1A Receptors. Med. Chem. Res. 1994, 4, 161. [Google Scholar]
- Paluchowska, M.H.; Bojarski, A.J.; Charakchieva-Minol, S.; Wesołowska, A. Active Conformation of Some Arylpiperazine Postsynaptic 5-HT1A Receptor Antagonists. Eur. J. Med. Chem. 2002, 37, 273. [Google Scholar]
- Wesołowska, A.; Paluchowska, M.H.; Gołębiowska, K.; Chojnacka-Wójcik, E. Pharmacological Characterization of MP 349, a Novel 5-HT1A Receptor Antagonist with Anxiolytic-Like Activity, in Mice and Rats. J. Pharm. Pharmacol. 2003, 55, 533. [Google Scholar]
- Griebel, G.; Rodgers, R.J.; Perrault, G.; Sanger, D.J. The Effects of Compounds Varying in Selectivity as 5-HT1A Receptor Antagonists in Three Rat Models of Anxiety. Neuropharmacology 2000, 39, 1848. [Google Scholar]
- Mokrosz, J.L.; Dereń-Wesołek, A.; Tatarczyńska, E.; Duszyńska, B.; Bojarski, A.J.; Mokrosz, M.J.; Chojnacka-Wójcik, E. 8-{4-[2-(1,2,3,4-Tetrahydroisoquinolinyl)]-butyl}-9-azaspiro[4.5]-decane-7,9-dione: A New 5-HT1A Receptor Ligand with the Same Activity Profile as Buspirone. J. Med. Chem. 1996, 39, 1125. [Google Scholar]
- Deren-Wesolek, A.; Tatarczynska, E.; Chojnacka-Wojcik, E. The novel buspirone analogue, 8-[4-[2-(1,2,3,4-tetrahydroisoquinolinyl)[butyl]-8-azaspiro [4.5]decane-7,9-dione, with anxiolytic-like and antidepressant-like effects in rats. J. Psychopharmacol. 1998, 12, 380. [Google Scholar]
- Paluchowska, M.H.; Mokrosz, M.J.; Bojarski, A.J.; Wesołowska, A.; Borycz, J.; Charakchieva-Minol, S.; Chojnacka-Wójcik, E. On the bioactive conformation of NAN-190 (1) and MP3022 (2), 5-HT(1A) receptor antagonists. J. Med. Chem. 1999, 42, 4952. [Google Scholar]
- Mokrosz, J.L.; Mokrosz, M.J.; Bojarski, A.J.; Charakchieva-Minol, S. Structure-Activity Relationship Studies of CNS Agents. Part 16: A Lower Limit of a Distance Between Crucial Pharmacophores of 5-HT1A Ligands. Pharmazie 1994, 49, 781. [Google Scholar]
- Mokrosz, M.J.; Bojarski, A.J.; Duszyńska, B.; Tatarczyńska, E.; Kłodzińska, A.; Dereń-Wesołek, A.; Charakchieva-Minol, S.; Chojnacka-Wójcik, E. 1,2,3,4-Tetrahydroisoquinoline derivatives: a new class of 5-HT1A receptor ligands. Bioorg. Med. Chem. 1999, 7, 287. [Google Scholar]
- Hackling, A.; Ghosh, R.; Perachon, S.; Mann, A.; Holtje, H.D.; Wermuth, C.G.; Schwartz, J.C.; Sippl, W.; Sokoloff, P.; Stark, H. N-(omega-(4-(2-methoxyphenyl)piperazin-1-yl)alkyl)carboxamides as dopamine D2 and D3 receptor ligands. J. Med. Chem. 2003, 46, 3883. [Google Scholar]
- Glennon, R.A.; Naiman, N.A.; Pierson, M.E.; Smith, J.D.; Ismaiel, A.M.; Titeler, M.; Lyon, R.A. N-(phthalimidoalkyl) derivatives of serotonergic agents: a common interaction at 5-HT1A serotonin binding sites? J. Med. Chem. 1989, 32, 1921. [Google Scholar]
- Bojarski, A.J.; Cegła, M.T.; Charakchieva-Minol, S.; Mokrosz, M.J.; Maćkowiak, M.; Mokrosz, J.L. Structure-Activity Relationship Studies of CNS Agents. Part 9: 5-HT1A and 5-HT2 Receptor Affinity of Some 2- and 3-Substituted 1,2,3,4-Tetrahydro-β-carbolines. Pharmazie 1993, 48, 289. [Google Scholar]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099. [Google Scholar]
- Sample Availability: Available from the authors.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Bojarski, A.J.; Mokrosz, M.J.; Duszyńska, B.; Kozioł, A.; Bugno, R. New Imide 5-HT1A Receptor Ligands – Modification of Terminal Fragment Geometry. Molecules 2004, 9, 170-177. https://doi.org/10.3390/90300170
AMA Style
Bojarski AJ, Mokrosz MJ, Duszyńska B, Kozioł A, Bugno R. New Imide 5-HT1A Receptor Ligands – Modification of Terminal Fragment Geometry. Molecules. 2004; 9(3):170-177. https://doi.org/10.3390/90300170
Chicago/Turabian StyleBojarski, Andrzej J., Maria J. Mokrosz, Beata Duszyńska, Aneta Kozioł, and Ryszard Bugno. 2004. "New Imide 5-HT1A Receptor Ligands – Modification of Terminal Fragment Geometry" Molecules 9, no. 3: 170-177. https://doi.org/10.3390/90300170