Investigations into the Use of a Polyfluorooctanol as an Auxiliary Component for an Aldol Reaction

Department of Chemistry, Queen Mary, University of London, London E1 4NS, UK
*
Author to whom correspondence should be addressed.
Molecules 2004, 9(5), 266-277; https://doi.org/10.3390/90500266
Submission received: 19 January 2004
/
Revised: 19 February 2004
/
Accepted: 12 March 2004
/
Published: 30 April 2004
(This article belongs to the Special Issue Carbanion chemistry from Carboxylic acids in honor of Prof. Ramón Mestres on his 65th anniversary)
Abstract
:Results are reported on the efficiency of polyfluorooctanol as a perfluorous auxiliary component in the aldol reaction between the enolate derived from polyfluorooctyl acetate and 2-fluorobenzaldehyde. Reduction of the corresponding polyfluoro β-hydroxy ester with Super Hydride® gave the required 1,3-diol in good yield.
Introduction
The direct synthesis of β-hydroxy carbonyl derivatives using an aldol reaction [1,2,3] by the addition of an enolisable carbonyl compound to an aldehyde (or ketone) is very well documented [4]. Over the last twenty five years, there have been many modifications of this reaction [5] most notably in the area of stereocontrol [6]. By comparison, very little attention has been paid to improving product separation within this reaction [7,8]. By far, the most common strategy has involved the use of a polymeric support attached to either the enolisable carbonyl derivative [9] or the aldehyde or ketone [10] to aid product separation and purification through simple filtration. This methodology is not perfect since it sometimes requires additional steps to couple or remove the required component from the polymeric support. Conceptually, a much simpler strategy would be one in which a single component could be isolated by selective removal from a multi-component mixture by using a less time consuming technique such as solvent extraction. This strategy has recently attracted a great deal of interest [11], most notably with the use of perfluorous solvents [12] which have been shown to selectively extract polyfluorinated components from non-fluorinated mixtures. This separation technique has been used to aid recovery of polyfluorous catalysts and auxiliaries [13]. More recently the use of fluorous silica gel to aid similar separations has been reported [14,15].
Results and Discussion
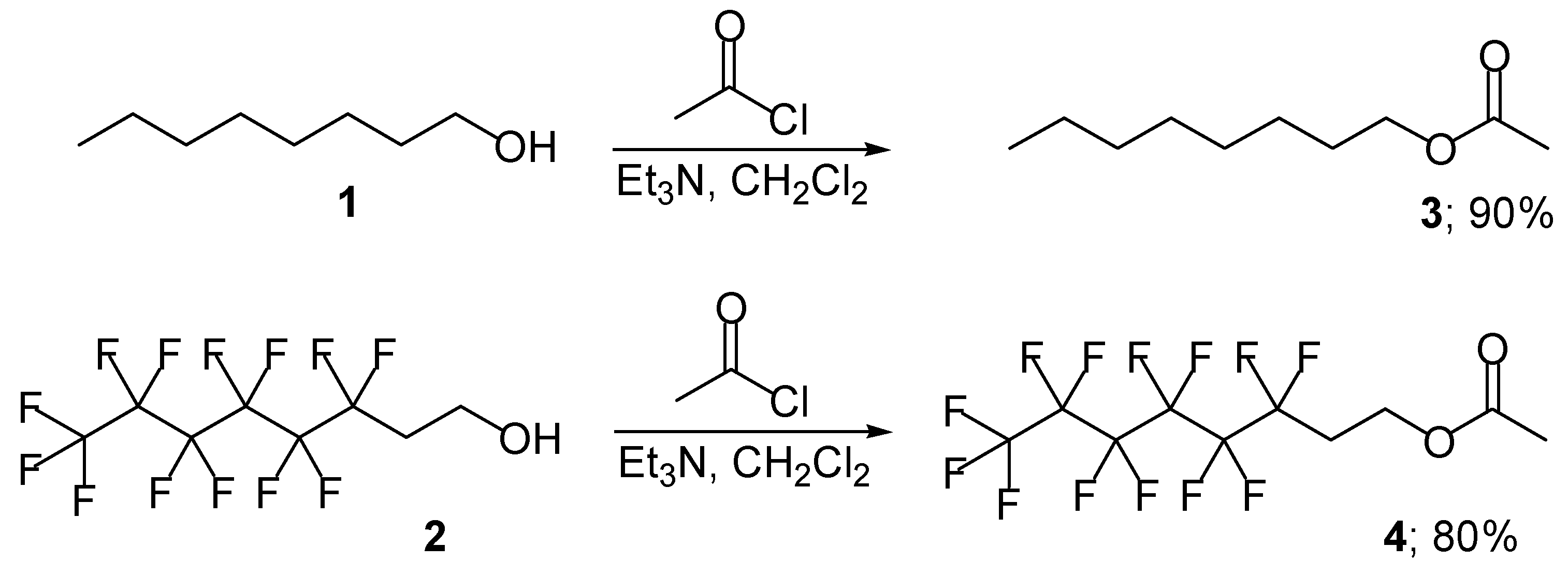
We now report our study into the use of polyfluorooctyl acetate 4 as an enolisable component for an acetate-based aldol reaction. For this study, we chose to synthesise the non-fluorinated and fluorinated octyl acetates 3 [16] and 4 to compare and contrast their differences as enolisable aldol components These acetates 3 and 4 were efficiently synthesised in high yield by the addition of octanol (1, [16]) and polyfluorooctanol 2, respectively, to a stirred solution of acetyl chloride and triethylamine in dichloromethane (Scheme 1).
Scheme 1.

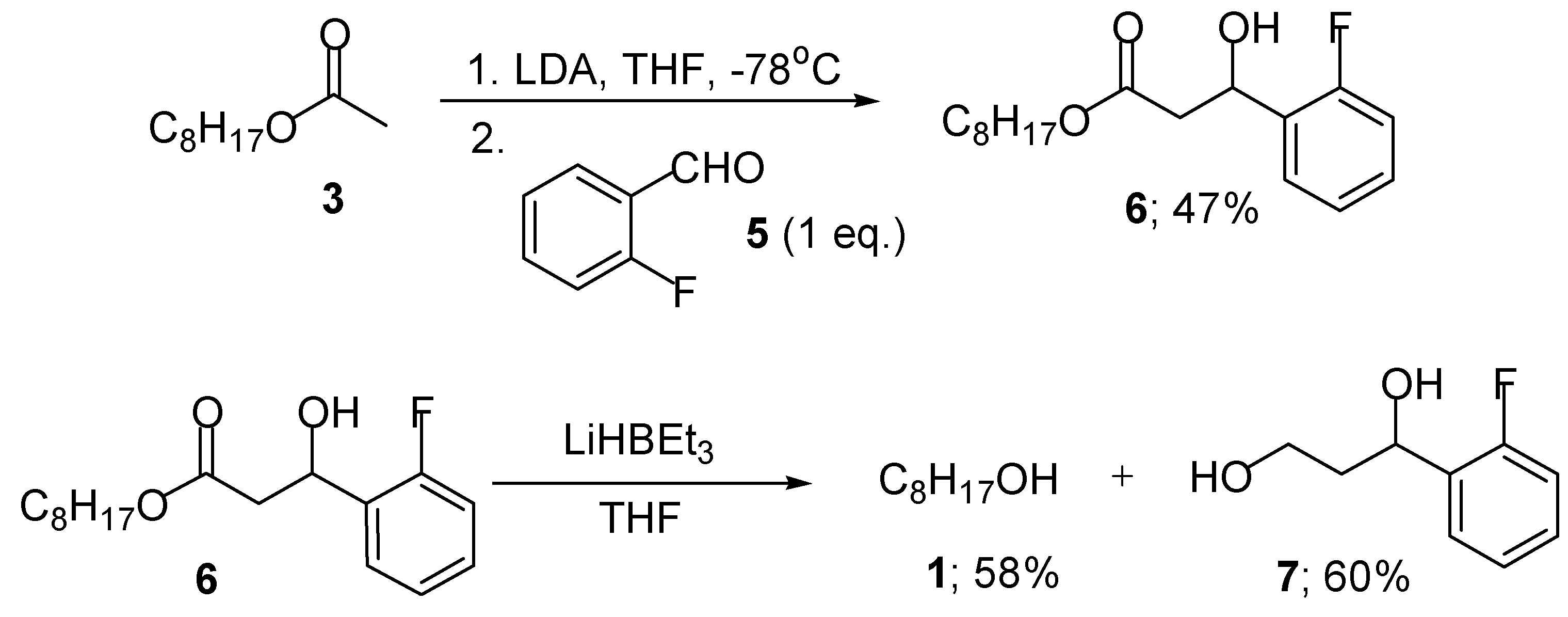
We first decided to investigate the aldol reaction between non-fluorinated octyl acetate 3 and 2-fluorobenzaldehyde (5) in an attempt to develop a standard protocol (Scheme 2). The corresponding lithium enolate of octyl acetate was formed by slow addition of lithium diisopropylamide (LDA) to a solution of 3 in THF at -78oC [17]. Addition of compound 5 to this solution gave the β-hydroxy ester 6 in moderate yield. Removal of the octanol component was easily achieved by simple reduction of the ester 6 with Super Hydride® to give the 1,3-diol 7 in 60% yield (Scheme 2).
Scheme 2.

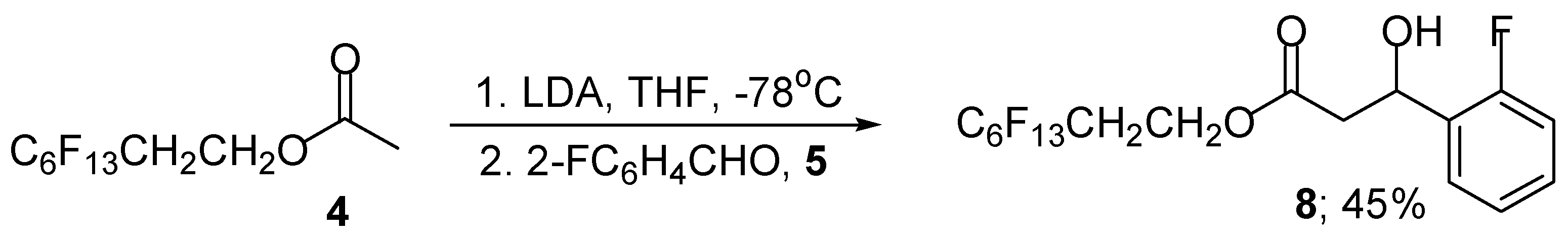
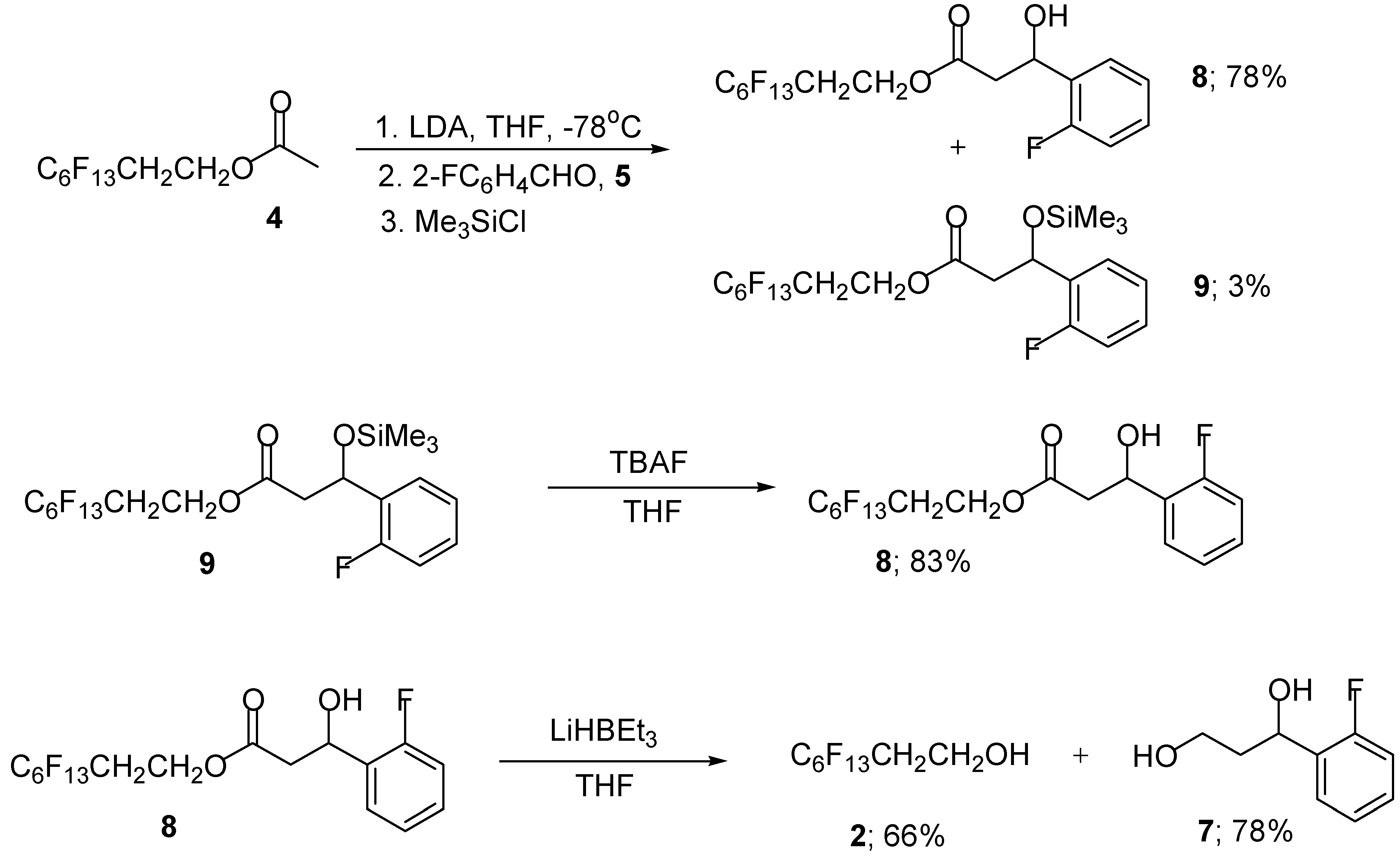
We next investigated the use of polyfluorooctyl acetate 4 as the enolisable component in this aldol reaction using our standard reaction conditions. Treatment of polyfluorooctyl acetate 4 with LDA, followed by the addition of 2-fluorobenzaldehyde (5) gave the required polyfluoro β-hydroxy ester 8 in moderate yield (Scheme 3). The yield could be improved significantly by the external addition of trimethylsilyl chloride (1 equivalent) to the intermediate aldolate to give, after flash column chromatography, the polyfluoro β-hydroxy ester 8 (78%) and silyl protected β-hydroxy ester 9 (3%) (Scheme 4).
Scheme 3.

The silyl protected ester 9 was easily converted to the required β-hydroxy ester 8 in 83% by simple addition of TBAF in THF (Scheme 4). Attempts at improving the yield of the silyl protected β-hydroxy ester 9 proved unsuccessful since it appears to be particularly unstable towards purification by flash column chromatography and preferentially fragments to give the parent β-hydroxy ester 8. Reduction of the polyfluoro β-hydroxy ester 8 proceeds smoothly to give the required 1,3-diol 7 (78%) and polyfluorooctanol 2 (66%) in good yield (Scheme 4).
Scheme 4.

With this information in hand, our attention next turned to the use of a perfluorous solvent such as FC-72 [18] (perfluorinated hexane - C6F14) to aid purification through selective extraction. We initially investigated the selective partition of our (non)-polyfluorinated derivatives 1-8 between an equal volume of FC-72 and THF [18] to see whether the use of a perfluorous solvent would aid product separation (Table 1). We were rather fortunate to find the polyfluoroooctyl acetate 4 and β-hydroxy ester 8 were selectively partitioned [19] into the perfluorous layer more so than their non-polyfluorinated derivatives 3 and 6 (Table 1). By comparison, the polyfluorooctanol 2 and polyfluorooctyl acetate 4 marginally favoured the more polar THF layer rather than the non-polar perfluorous FC-72 layer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
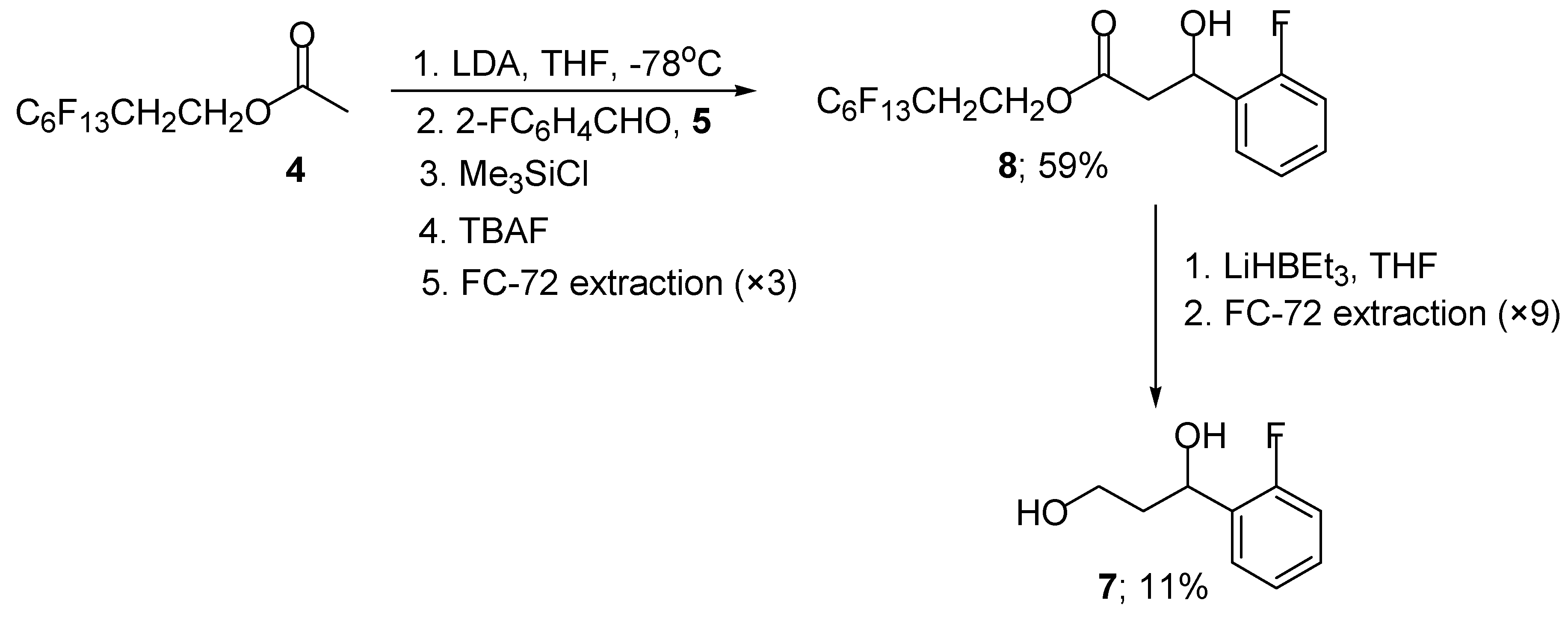{kind=link}
{kind=link}
| THF/FC-72 | 98/2 | 70/30 | 95/5 | 67/33 | 97/3 | 92/8 | 97/3 | 21/79 | ||||||
| THF/FC-72 | 5/95 | 95/5 | 83/17 | 86/14 | 95/5 | 74/26 | 92/8 | |||||||
From inspection of these partition ratios, it appears that the use of perfluorous extraction could aid purification in our original aldol reaction since the product, polyfluoro β-hydroxy ester 8 would favour the perfluorous layer (FC-72), whereas, the starting precursors, polyfluorooctyl acetate 4 and 2-fluorobenzaldehyde (5) would favour the organic (THF) layer. Thus, by repeating the original aldol reaction between polyfluorooctyl acetate 4 and compound 5, and extracting the crude reaction residue [20] three times with FC-72 and THF the required β-hydroxy ester 8 was obtained in 59% yield and in high purity (>95% - determined by 1H NMR spectroscopy) (Scheme 5).
Scheme 5.

Simple reduction of the polyfluoro β-hydroxy ester 8 with Super Hydride® gave the required 1,3-diol 7. Perfluorous extraction of the crude reaction residue [17] was performed nine times with a combination of FC-72 and THF to allow the 1,3-diol 7 to be recovered in high purity (>95% - determined by 1H-NMR spectroscopy). The recoverability of 1,3-diol 7 was evidently lower than that obtained by purification by flash chromatography (11% versus 88%) due to the large number of perfluorous extractions required to separate the diol 7 from the polyfluorooctanol component 2.
Scheme 6.

An attempt at extending this methodology towards other aromatic aldehydes like 10 and 13 proved more difficult due to a lower perfluorous partition ratio (Table 1) for the aldol adducts 11 and 14 compared to 8 (Scheme 6). The presence of the 2-fluoro substituent on the aromatic aldehyde 5 appears to be important; this may presumably be due to a lowering the polarity of the aldol adduct 8 by the 2-fluoro substituent, thus promoting its partition into the FC-72 layer more so than related aldol adducts 11 and 14 [21].
Conclusions
In conclusion, we have shown that polyfluorooctanol can be used as a perfluorous auxiliary component within an aldol reaction. The polyfluorooctanol auxiliary 2 can efficiently be removed by reduction of the β-hydroxy ester 8 with Super Hydride® to give a synthetically useful 1,3-diol 7. The yields for the isolation of pure β-hydroxy ester 8 and 1,3-diol 7 were found to be higher when using column chromatography to purify the crude reaction mixtures than using selective perfluorous extraction. This was primarily due to the need for additional perfluorous extractions to ensure removal of the unwanted byproducts.
Experimental
General
All solvents were distilled before use. Tetrahydrofuran (THF) and ether were freshly distilled from LiAlH4. Triphenylmethane was used as the indicator for THF. All reactions were carried out under nitrogen using oven-dried glassware. Flash column chromatography was carried out using Merck Kieselgel 60 (230-400 mesh). Thin layer chromatography (TLC) was carried out on commercially available pre-coated plates (Merck Kieselgel 60F254 silica). A 1:1 light petroleum (40-60oC)-ether solvent system was used for elution in both techniques. 1H- and 13C-NMR spectra were recorded on a JEOL EX 270 and Bruker AM 250, AMX 400 and AM 600 Fourier transform spectrometers (using an internal deuterium lock). Chemical shifts are quoted in parts per million downfield from tetramethylsilane. 13C-NMR spectra were recorded with broad proton decoupling. Infrared spectra were recorded on a Shimadzu 8300 FTIR instrument and mass spectra were recorded on a Kratos 50MSTC spectrometer using a DS503 data system for high-resolution analysis.
Octyl acetate (3) [16]
Acetyl chloride (2.48 g, 31.6 mmol) was slowly added to a stirred solution of octanol (1) (4.12 g, 31.6 mmol) and Et3N (3.19 g, 31.6 mmol) in a CH2Cl2 (100 mL) at 25 °C. The reaction mixture was stirred for 12 hours. Water (50 mL) was added and the mixture was extracted with dichloromethane (2 ° 30 mL). The combined organic layers were dried (MgSO4) and evaporated under reduced pressure. The residue was purified by flash column chromatography to give octyl acetate (3) (4.91 g, 90 %) as colourless oil; RF 0.75; IR vmax(KBr)/cm-1 2927 (C-H), 1743 (C=O) and 1039 (CO); 1H-NMR δH (250 MHz, CDCl3) 4.08 (2 H, t, J 6.6, CH2O), 2.01 (3 H, s, COCH3), 1.60 (2 H, m, CH2CH2O), 1.31-1.10 (10 H, m, 5 × CH2) and 0.96 (3 H, t, J 6.6, CH3CH2); 13C-NMR δC (62.8 MHz, CDCl3) 171.2 (C=O), 64.6 (CH2O), 31.8, 29.2, 28.6, 25.9, 22.6 and 21.0 (6 × CH2), 17.5 (COCH3) and 14.1 (CH3CH2); HRMS: Found M + H, 173.2779, C10H20O2 requires M+, 172.2725.
Polyfluorooctyl acetate (4)
Polyfluorooctanol (2) (5.21 g, 14.29 mmol) was added to a stirred solution of acetyl chloride (1.12 g, 14.29 mmol) and Εt3Ν (1.44 g, 14.29 mmol) in CH2Cl2 (100 mL) at room temperature. The reaction mixture was stirred for 12 hours. Water (50 mL) was added and the mixture was extracted with dichloromethane (2 ° 30 mL). The combined organic layers were dried (MgSO4) and evaporated under reduced pressure. The residue was purified by flash column chromatography to give the polyfluorooctyl acetate (4) (5.50 g, 78 %) as a colourless solution; RF 0.85; vmax(KBr)/cm-1 2995 (C-H), 1751 (C=O) and 1022 (CO); 1H-NMR δH (250MHz, CDCl3) 4.35 (2 H, t, J 6.6, CH2O), 2.45 (2 H, m, CF2CH2CH2) and 2.04 (3 H, s, COCH3); δC (62.8 MHz, CDCl3) 178.4 (C=O), 127.5, 120.15, 112.4, 110.8, 109.3, and 106.7 (6 × CF2), 56.2 (CH2O), 30.5 (CF3CH2) and 20.3 (COCH3); HRMS: Found M+ 363, C10H7O2F13 requires M +, 406.0238.
Octyl 3-hydroxy-3- (2’-fluorophenyl) propionate (6)
Octyl acetate (3) (4.07 g, 5.05 ml, 24.22 mmol) was added to a solution of lithium diisopropylamide (16.1 mL, 1.5 M in THF, 24.22 mmol) in freshly distilled THF (20 mL) at -78 °C and the resulting solution was stirred for 45 minutes. A solution of 2-fluorobenzaldehyde (5) (3.00 g, 2.55 mL, 24.22 mmol) in THF (10 mL) was added dropwise and the resulting solution was stirred for 3 hours. The reaction was quenched with water (50 mL) and the mixture was extracted with ether (3 ° 50 mL). The combined organic layers were dried (MgSO4) and evaporated under reduced pressure. The residue was purified by flash column chromatography to give the ester 6 (3.30 g, 47 %) as a colourless oil; RF 0.58; vmax (KBr)/cm-1 3373 (O-H), 2925 (C-H), 1735 (C=O), 1616 (C=C) and 1070 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.85-6.94 (4 H, m, 4 × CH; Ar), 5.46-5.36 (1 H, dd, J 8.2 and 4.6, CHOH), 4.12 (2 H, t, J 6.6, OCH2), 3.22 (1 H, br s, OH), 2.70-2.60 (2 H, m, CH2) 1.57 (2 H, m, CH2CH2O), 1.30-1.10 (10 H, m, 5 × CH2) and 0.96 (3 H, t, J 6.6, CH3CH2); 13C-NMR: δC (62.8 MHz, CDCl3) 172.3 (C=O), 147.2 (i-CF; Ar), 139.8 (i-C-C; Ar), 133.7 and 124.5 (m-C; Ar), 128.3 (p-C; Ar), 128.2 (o-C; Ar), 66.7 (CHOH), 61.6 (CH2O), 42.3 (CH2C=O), 32.5, 30.0, 29.9, 28.6, 26.5 and 23.1 (6 × CH2), and 14.1 (CH3); HRMS: Found M+, 297.3885, C17H25O3F requires M+, 297.3808.
3-Hydroxy-3-(2’-fluorophenyl)propanol (7)
Super Hydride® (LiBHEt3) (3.91 mL, 1.0 M in THF, 12.9 mmol) was added slowly to a stirred solution of the aldol adduct (6) (1.15 g, 3.91 mmol) in THF (10 mL) at room temperature. The resulting solution was stirred for 2 hours and quenched with water (5 mL). The solution was partitioned between ether and water (50 mL) and the organic layer was extracted with ether (2 × 25 mL). The combined organic layers were dried (with magnesium sulphate), filtrated and evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel to give octanol 1 (29 mg, 58 %) as a colourless liquid and the diol 7 (0.39 g, 60 %) as a colourless oil; RF 0.38; IR vmax(KBr)/cm-1 3461 (O-H), 2881 (C-H), 1585 (C=C) and 1080 (C-O); 1H-NMR δH (250 MHz, CDCl3) 7.48-6.98 (4 H, m, 4 × CH; Ar), 5.35 (1 H, dd, J 5.6 and 3.4 CHOH), 4.14-3.94 (2 H, m, CH2OH), 3.65 (1 H, br s, OH) and 2.30-1.80 (2 H, m, CH2); 13C-NMR δC(62.8 MHz, CDCl3) 161.4 (i-CF; Ar), 157.5 (i-C; Ar), 129.9 (o-C; Ar), 126.7 (p-C; Ar), 124.3 and 115.4 (m-C; Ar), 67.4 (CHOH), 61.4 (CH2OH) and 41.4 (CH2CH2OH); HRMS: Found M+, 167.1604, C9H11O2 F requires M, 170.1841.
Polyfluorooctyl 3-hydroxy-3- (2’-fluorophenyl) propionate (8)
In the same way as described for the ester 6, lithium diisopropylamide (2.0 mL, 1.5 M in THF, 3.0 mmol), polyfluorooctyl acetate (4) (0.95 g, 2.35 mmol), 2-fluorobenzaldehyde (5) (0.29 g, 0.25 mL, 2.35 mmol) and Me3SiCl (0.25 g, 0.30 ml, 2.3 mmol) in THF (50 mL) gave, after flash chromatography the perfluorous ester 8 (0.97 g, 78%) as a colourless oil; RF 0.48; IR vmax (KBr)/cm-1 3373 (O-H), 2925 (C-H), 1735 (C=O) 1585 (C=C) and 1070 (C-O); 1H-NMR δH(250 MHz, CDCl3) 7.55- 6.95 (4 H, m, 4 × CH; Ar), 5.46-5.41 (1 H, dd, J 7.9 and 4.6, CHOH), 4.45 (2 H, t, J 6.5, OCH2), 3.35 (1 H, br s, CHOH), 2.83-2.45 (2 H, m, CH2) and 2.60-2.35 (2 H, m, OCH2CH2CF2); 13C-NMR δC(62.8 MHz, CDCl3) 171.7 (C=O), 161.4 (i-CF; Ar), 157.5 (i-CC; Ar), 129.3 (p-C; Ar), 127.5 (o-C; Ar), 127.2, 120.15, 112.4, 110.8, 109.3 and 106.7 (6 × CF2), 61.6 (CHOH), 56.6 (CH2O), 37.6 (O=COCH2) and 22.4 (CF2CH2); HRMS: Found M + H, 513.261, C17H12O3F14 requires M, 530.2533); and silyl protected ester (9) (42 mg, 3%) as a colourless oil; RF 0.86; IR vmax (KBr)/cm-1 3469 (O-H), 2922 (C-H), 1752 (C=O), 1614 (C=C) and 1083 (C-O); 1H-NMR δH (250 MHz, CDCl3) 7.56-6.98 (4 H, m, 4 × CH; Ar), 5.47 (1 H, dd, J 7.8 and 4.6, CHOSiMe3), 4.48 (2 H, t, J 6.6, OCH2), 2.85-2.46 (2 H, m, CH2), 2.80-2.40 (2 H, m, CH2CF2) and 0.09 (9 H, s, Si(CH3)3); 13C-NMR δC (62.8 MHz, CDCl3) 172.1 (C=O), 162.3 (i-C-F; Ar) 140.9 (i-C; Ar) 128.7 (m-C; Ar), 127.4 (p-C; Ar), 127.3 (o-C; Ar), 120.7, 119.8, 110.8, 106.2, 105.2, and 101.9 (6 × CF2), 68.4 (COSiMe3), 56.8 (CH2O), 44.3 (O=CCH2) and 25.9 (CH2CF2); HRMS: Found M, 593.3622, C20H19O3F14Si requires M, 601.4254.
Synthesis of 3-hydroxy-3-(2’-fluorophenyl)propanol (7) derived from octyl 3-hydroxy-3-(2’-fluoro-phenyl)propionate (8)
In the same way as diol 7, the polyfluoro ester 8 (0.17 g, 0.32 mmol) and Super Hydride® (1.06 mL, 1.0 M in THF, 1.06 mmol) in THF (50 mL) gave, after flash column chromatography, a separable mixture of polyfluorooctanol 2 (64 mg, 66 %) and diol 7 (47 mg, 88 %) both as colourless oils; RF 0.40, 0.38; all compounds were spectroscopically identical to those obtained previously.
Purification by perfluorous extraction using THF/FC-72
Synthesis of polyfluorooctyl 3-hydroxy-3- (2’-fluorophenyl) propionate (8)
Polyfluorooctyl acetate 4 (1.16 g, 2.86 mmol) was added to a stirred solution of lithium diisopropylamide (1.91 mL, 1.5 M in THF, 3.55 mmol) in THF at –78 °C. The resulting solution was stirred for 45 minutes. 2-Fluorobenzaldehyde (5) (0.29 g, 0.25 mL, 2.35 mmol) was added dropwise to this solution and stirred for a further 10 minutes. Me3SiCl (0.15 mL, 0.18 ml, 2.86 mmol) was slowly added and the resulting mixture was stirred for a further 4 hours. The reaction was quenched by the addition of dilute HCl (3 M, 2 mL) followed by the addition of water (50 mL). The organic layer was extracted with ether (3 × 25 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (2 × 25 mL), dried (with MgSO4) and evaporated under reduced pressure. By TLC this residue was shown to contain both the required polyfluoro ester 8 and the silyl protected ester 9. This residue was converted to the ester 8 by addition of tetrabutylammonium fluoride (TBAF) (4 equivalents) in THF (10 mL) and stirring overnight. The reaction mixture was quenched with water (20 mL) and extracted with ether (2 × 25 mL) and the combined organic layers were dried with MgSO4. This residue was purified by three extractions using THF (10 mL) and FC-72 (10 mL). By 1H-NMR of the FC-72 extract contained only the polyfluorinated ester 8 (0.73 g, 59 %) and was shown to be spectroscopically identical to that obtained previously.
Synthesis of 3-hydroxy-3-(2’-fluorophenyl)propanol (7)
Super Hydride® (1.06 mL, 1.0 M in THF, 1.06 mmol) was added slowly to a stirred solution of the polyfluoro ester 8 (0.17 g, 0.32 mmol) in THF (10 mL) at 0 °C. The reaction mixture was stirred for 2 hours. Water (10 mL) was added and the solution was extracted with ether (2 ° 25 mL). The combined organic layers were dried (MgSO4) and evaporated under reduced pressure. The mixture was separated and purified by nine extractions using THF (5 mL) and FC-72 (5 mL). The required 1,3 diol 7 was isolated from the organic (THF) phase (6 mg 11%) and was shown to be spectroscopically identical to that obtained previously.
Polyfluorooctyl 3-hydroxy-3-phenyl propionate (11)
In the same way as for the ester 6, lithium diisopropylamide (2.37 mL, 1.5 M in THF, 3.55 mmol), perfluorooctyl acetate (4) (1.12 g, 2.75 mmol), benzaldehyde (10) (0.29 g, 0.28 mL, 2.75 mmol) and Me3SiCl (0.29 g, 0.34 mL, 2.75 mmol) in THF (50 mL) gave, after flash chromatography on silica gel the polyfluoro ester 11 (0.82 g, 58 %) as an oil; RF 0.35; IR vmax (KBr)/cm-1 3469 (OH), 2921 (CH), 1745 (C=O), 1652 (C=C) and 1070 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.54-7.22 (5 H, m, 5 × CH; Ar), 5.37 (1 H, ddd, J 8.2, 4.9 and 3.6, CHOH), 4.95 (2 H, t, J 6.6, OCH2), 3.45 (1 H, br s, OH), 2.75-2.40 (2 H, m, CH2) and 2.20 (2 H, m, CF2CH2CH2O); 13C-NMR δC (62.8 MHz, CDCl3) 163.4 (C=O), 132.1 (i-C; Ar), 119.4 (p-C; Ar), 119.3 (m-C; Ar), 117.6 (o-C; Ar), 111.3, 109.7, 108.2, 106.7, 102.8, and 102.1 (6 × CF2), 63.6 (CHOH), 48.0 (CH2O), 37.6 (O=CCH2) and 27.6 (CF2CH2CH2); HRMS: Found M + H, 512.0683, C17H13O3F13 requires M, 512.0657; MS: m/z 495.3 (45%, M - H2O + H) and 453.0 (100, M – C2H3O2); and the corresponding silyl protected ester (0.32 g, 19 %) as a colourless oil; RF 0.86; IR vmax (KBr)/cm-1 3451 (OH), 2927 (CH),1730 (C=O), 1622 (C=C) and 1060 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.50-7.10 (5 H, m, 5 × CH; Ar), 5.19 (1 H, dd, J 8.2 and 4.4, CHOSiMe3), 4.10 (2 H, t, J 6.6, OCH2), 2.75-2.40 (2 H, m, CH2), 1.65 (2 H, m, OCH2CH2CF2) and 0.09 (9 H, s, SiMe3); 13C-NMR δC (62.8 MHz, CDCl3) 172.0 (C=O), 140.9 (i-C, Ar), 128.7 (m-C; Ar), 127.4 (p-C, Ar),127.3 (o-C, Ar), 120.0, 119.2, 110.2, 106.2, 105.2, and 101.9 (6 × CF2), 68.4 (COSiMe3), 53.1 (CH2O) 44.3 (O=CCH2), and 25.9 (CF2CH2); HRMS: Found M+, 584.1033. C20H21F13O3Si requires M, 584.1052.
3-Hydroxy-3-phenyl propanol (12) [22]
In the same way as described for diol 7, the polyfluorous ester 11 (0.23 g, 0.44 mmol) and Super Hydride® (2.5 mL, 1.0 M in THF, 2.5 mmol) in THF (20 mL) gave, after flash column chromatography, a separable mixture of polyfluoro octanol 2 (64 mg, 40 %) and diol 12 (41 mg, 61 %) as colourless oil; RF 0.33; IR vmax(KBr)/cm-1 3388 (OH), 2968 (CH) and 1060 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.41-7.26 (5 H, m, 5 × CH; Ph), 5.10 (1 H, dd, J 5.6 and 3.5, CHOH), 4.26-3.81 (2 H, m, CH2OH), 2.27-1.86 (2 H, m, CH2) and 1.30 (1 H, br s, OH); 13C-NMR δC (62.8 MHz, CDCl3) 138.5 (i-C; Ph), 128.5 (o-C; Ph), 127.5 (m-C; Ph), 125.2 (p-C; Ph), 72.5 (CHOH), 60.7 (CO) and 35.2 (CH2CH2OH); HRMS: Found M+, 152.176. C9H12O2 requires M, 152.1768; MS: m/z 152.2 (22%, M) and 105.2 (100, M - CH2CH2OH).
Polyfluorooctyl 3-hydroxy-3- (2’-methylphenyl) propionate (14)
In the same way as for the ester 6, lithium diisopropylamide (1.77 mL, 1.5 M in THF, 2.65 mmol), polyfluorooctyl acetate (4) (1.08 g, 2.65 mmol), o-tolualdehyde (13) (0.46 g, 2.65 mmol) and Me3SiCl (0.28 g, 0.33 mL, 2.65 mmol) in THF (50 mL) gave, after flash chromatography, the ester 14 (1.23 g, 81 %) as a colourless oil; RF 0.44; IR vmax (KBr)/cm-1 3469 (OH), 2921 (CH), 1745 (C=O), 1612 (C=C) and 1070 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.54-7.22 (4 H, m, 4 × CH; Ar), 5.37 (1 H, ddd, J8.2, 4.4, and 3.4; CHOH), 4.95 (2 H, t, J 6.6, OCH2), 3.45 (1 H, br s, OH), 2.75-2.40 (2 H, m, CH2), and 2.20 (2 H, br s CF2CH2CH2O); 13C-NMR δC(62.8 MHz, CDCl3) 163.7 (C=O), 132.1 (i-C-C; Ar), 126.1 (i-C-CH3; Ar), 122.3 (o-C; Ar), 119.5 and 119.3 (m-C; Ar), 119.4 (p-C; Ar), 120.0, 119.2, 110.2, 106.2, 105.2 and 101.9 (6 × CF2), 58.8 (CHOH), 48.9 (CH2O), 41.5 (O=CCH2) and 33.9 (CH2CF2); HRMS: Found M + H, 526.2903, C17H13O3F13 requires M, 526.2885; MS: m/z 509.3 (30%, M - H2O + H) and 467.2 (100, M - C2H3O2); and the corresponding silyl protected ester (0.11 g, 7 %) as a colourless oil; RF 0.73; IR vmax (KBr)/cm-1 3473 (OH), 2976 (CH), 1749 (C=O), 1602 (C=C) and 1082 (CO); 1H-NMR δH (250 MHz, CDCl3) 7.53-7.21 (4 H, m, 4 × CH; Ar), 5.35 (1 H, dd, J 8.2, and 4.5, CHOSiMe3), 4.93 (2 H, t, J 6.6, OCH2), 2.65-2.40 (2 H, m, CH2), 1.89 (2 H, m, OCH2CH2CF2) and 0.09 (9 H, s, SiMe3); 13C-NMR δC (62.8 MHz, CDCl3) 172.0 (C=O), 140.9 (i-C; Ar), 128.7 (p-C; Ar), 127.4 (m-C; Ar), 127.3 (o-C; Ar), 120.7, 119.8, 110.8, 106.2, 105.2, and 101.9 (6 × CF2), 68.4 (COSiMe3), 53.1 (CH2O) and 25.9 (CH2CF2); HRMS: Found M+, 598.1256. C21H23F13O3Si requires M, 598.1209.
3-Hydroxy-3-(2’-methylphenyl)propanol (15)
In the same way as for diol 7, the polyfluoro ester 14 (1.03 g, 1.95 mmol) and Super Hydride® (3.3 mL, 1.0 M in THF, 3.30 mmol) in THF (20 mL) gave, after flash column chromatography, a separable mixture of polyfluorooctanol 2 (0.26 g, 37 %) and diol 15 (0.18 g, 55 %) as colourless oil; RF 0.55; vmax(KBr)/cm-1 3462 (OH), 2879 (CH), 1080 (CO) and 723 (aromatic; ortho-disubstituted); 1H-NMR δH(250 MHz, CDCl3) 7.40-7.20 (5 H, m, 5 × CH; Ph), 5.26 (1 H, dd, J 5.6 and 3.5, CHOH), 4.10-3.98 (2 H, m, CH2OH), 2.30 (3 H, s, OCH3; Ar), 2.28-1.20 (2 H, m, CH2) and 1.35 (1 H, br s, OH); 13C-NMR δC(62.8 MHz, CDCl3) 140.5 (i-C; Ph), 133.8 (i-CCH3; Ph), 130.5 (m-C; Ph), 127.4 (o-C; Ph), 126.5 (p-C; Ph), 124.1 (m-C; Ph), 70.1 (CHOH), 60.9 (C-O), 33.7 (CH3; Ar) and 18.8 (CH2CH2OH); HRMS: Found M+ - H, 165.2125, C10H14O2 requires M, 166.2194; MS: m/z 130.2 (100, M - 2 × H2O).
References and Notes
- Heathcock, C.H. Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Volume 2, p. 133, and references therein. [Google Scholar]
- Heathcock, C.H. Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Volume 2, p. 181. [Google Scholar]
- Mukaiyama, T. Org. React. 1982, 28, 203.
- For recent examples involving acetate aldols, see: Arefolov, A.; Panek, J.S. Org. Lett. 2002, 4, 2397–2400.Guz, N.R.; Phillips, A.J. Org. Lett. 2002, 4, 2253–2256.Singer, R.A.; Shepard, M.S.; Carreira, E.M. Tetrahedron 1998, 54, 7025–7032.Denmark, S.E.; Winter, S.B.D. Synlett 1997, 1087–1089.
- Palomo, C.; Oiarbide, M.; García, J.M. Chem. Eur. J. 2002, 8, 37–44.
- Paterson, I. Pure Appl. Chem. 1992, 64, 1821–1830.Cowden, C.J.; Paterson, I. Org. React. 1997, 51, 1.
- Studer, A.; Hadida, S.; Ferritto, R.; Kim, S.Y.; Jeger, P.; Widf, P.; Curran, D.P. Science 1997, 275, 823–826.
- Tzschucke, C.C.; Markert, C.; Bannwarth, W.; Roller, S.; Hebel, A.; Haag, R. Angew. Chem. Int. Ed. Engl. 2002, 41, 3964–4000.
- Phoon, C.W.; Abell, C. Tetrahedron Lett. 1998, 39, 2655–2658.Kobayashi, S.; Hachiya, I.; Yasuda, M. Tetrahedron Lett. 1996, 37, 5569–5572.
- Reggelin, M.; Brenig, V. Tetrahedron Lett. 1996, 37, 6851–6852.
- Gladysz, J.A.; Curran, D.P. Tetrahedron 2002, 58, 3823–3825.Kitazume, T. J. Fluorine Chem. 2000, 105, 265–278.Curran, D.P.; Lee, Z. Green Chem. 2001, G3.
- Dobbs, A.P.; Kimberley, M.R. J. Fluorine Chem. 2002, 118, 3–17, and references therein.
- Some recent examples include: Bayardon, J.; Cavazzini, M.; Maillard, D.; Pozzi, G.; Quici, S.; Sinou, D. Tetrahedron: Asymmetry 2003, 13, 2215–2224.Adams, D.J.; Gudmunsen, D.; Fawcett, J.; Hope, E.G.; Stuart, A.M. Tetrahedron 2002, 58, 3827–3834.Dandapani, S.; Curran, D.P. Tetrahedron 2002, 58, 3855–3864.rich, D.; Neelamkavil, S. Tetrahedron 2002, 58, 3865–3870.Itsuno, S.; Komura, K. Tetrahedron 2002, 58, 8237–8246.Morphy, J.R.; Rankovic, Z.; York, M. Tetrahedron Lett. 2002, 43, 6413–6415.Nakamura, Y.; Takeuchi, S.; Ohgo, Y. J. Fluorine Chem. 2003, 120, 121–129.Mikami, K.; Mikami, Y.; Matsumoto, Y.; Nishikido, J.; Yamamoto, F.; Nakajima, H. Tetrahedron Lett. 2001, 42, 289–292.
- Zhang, W.; Curran, D.P.; Chen, C.H.T. Tetrahedron 2002, 58, 3871–3875.
- Zhang, W. Tetrahedron 2003, 59, 4475–4489.
- Ishihara, K.; Kurihara, H.; Yamamoto, H. J. Org. Chem. 1993, 58, 3793–3791.
- Related β-hydroxy esters derived from octyl acetate have previously been prepared using a Reformatsky reaction. See: Chollet, J.-F.; Miginiac, L.; Picotin, G.; Miginiac, P. Synth. Commun. 1989, 19, 2167–2173.
- Nakamura, Y.; Takeuchi, S.; Ohgo, Y.; Curran, D.P. Tetrahedron 2000, 56, 351–356.
- A mixture of 100 mg of substrate was added to a solution of CF-72 (2 mL) and THF (2 mL). The two phases were separated and the solvents were evaporated under vacuum. The contents of the CF-72 and THF layers were determined by weighing the residue (for further information - see reference 18).
- The crude reaction mixture (after separation and extraction with ether and water) was added to a solution of CF-72 (10 mL) and THF (10 mL). The two phases were separated and the solvents were evaporated under vacuum. The contents in the THF layer was extracted under the conditions outlined above.
- Alcohols (e.g., 1) prefer the THF layer rather than the FC-72 layer due to their polar nature. However, if polarity of the alcohol is decreased (e.g., 2) the FC-72 partition ratio could presumably be increased (cf. Table 1). The polarity of the OH group in 6, 8, 11 and 14 may be lower than expected due to internal hydrogen bonding [to the adjacent carbonyl (C=O) group (e.g., 6, 8, 11 and 14) or the 2'-fluorophenyl substituent (e.g., 8)]. The polarity of the OH group (in 8) can be lowered further by forming the corresponding non-polar silyl ether (e.g., 9) which promotes partitioning into the FC-72 layer rather than the THF layer (Table 1).
- Kiyooka, S.; Kaneko, Y.; Komura, M.; Matsuo, H.; Nakano, M. J. Org. Chem. 1991, 56, 2276–2278.
- Sample availability: Contact the authors.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Eames, J.; Khanom, H. Investigations into the Use of a Polyfluorooctanol as an Auxiliary Component for an Aldol Reaction. Molecules 2004, 9, 266-277. https://doi.org/10.3390/90500266
AMA Style
Eames J, Khanom H. Investigations into the Use of a Polyfluorooctanol as an Auxiliary Component for an Aldol Reaction. Molecules. 2004; 9(5):266-277. https://doi.org/10.3390/90500266
Chicago/Turabian StyleEames, Jason, and Hasina Khanom. 2004. "Investigations into the Use of a Polyfluorooctanol as an Auxiliary Component for an Aldol Reaction" Molecules 9, no. 5: 266-277. https://doi.org/10.3390/90500266