Theoretical Studies on Electronic States of Rh-C60. Possibility of a Room-temperature Organic Ferromagnet

Abstract
:Introduction

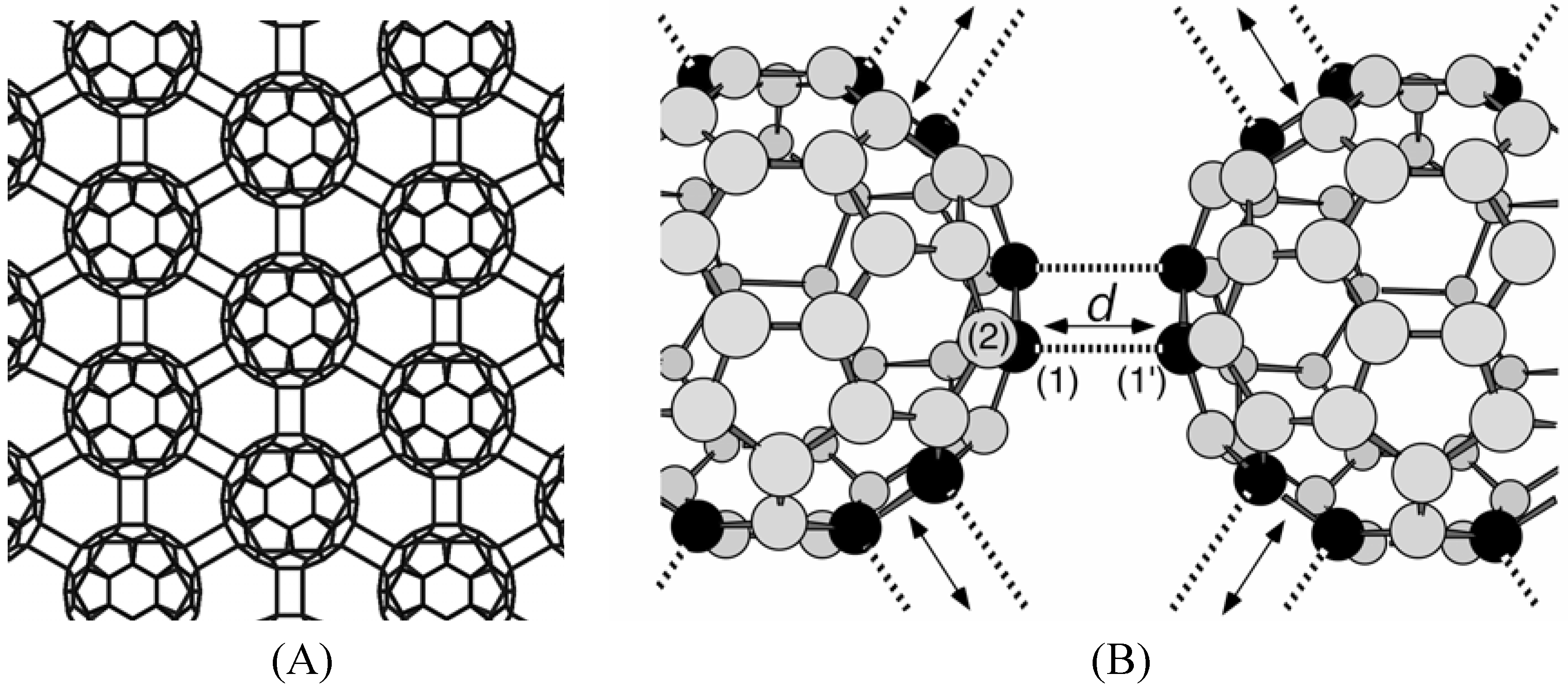
Experimental background

Computational details
sp2-sp3 rehybridization

| Model | C*(1)-C(2) | C*(1)-C*(1') |
|---|---|---|
| C60 (R-3m) | 0.459 | 0.008 |
| C60 (Fm-3) | 0.427 | 0.005 |
| Graphite | 0.440 | |
| Diamond | 0.319 | |
| Rh-C60 (d=8.3%) | 0.417 | 0.013 |
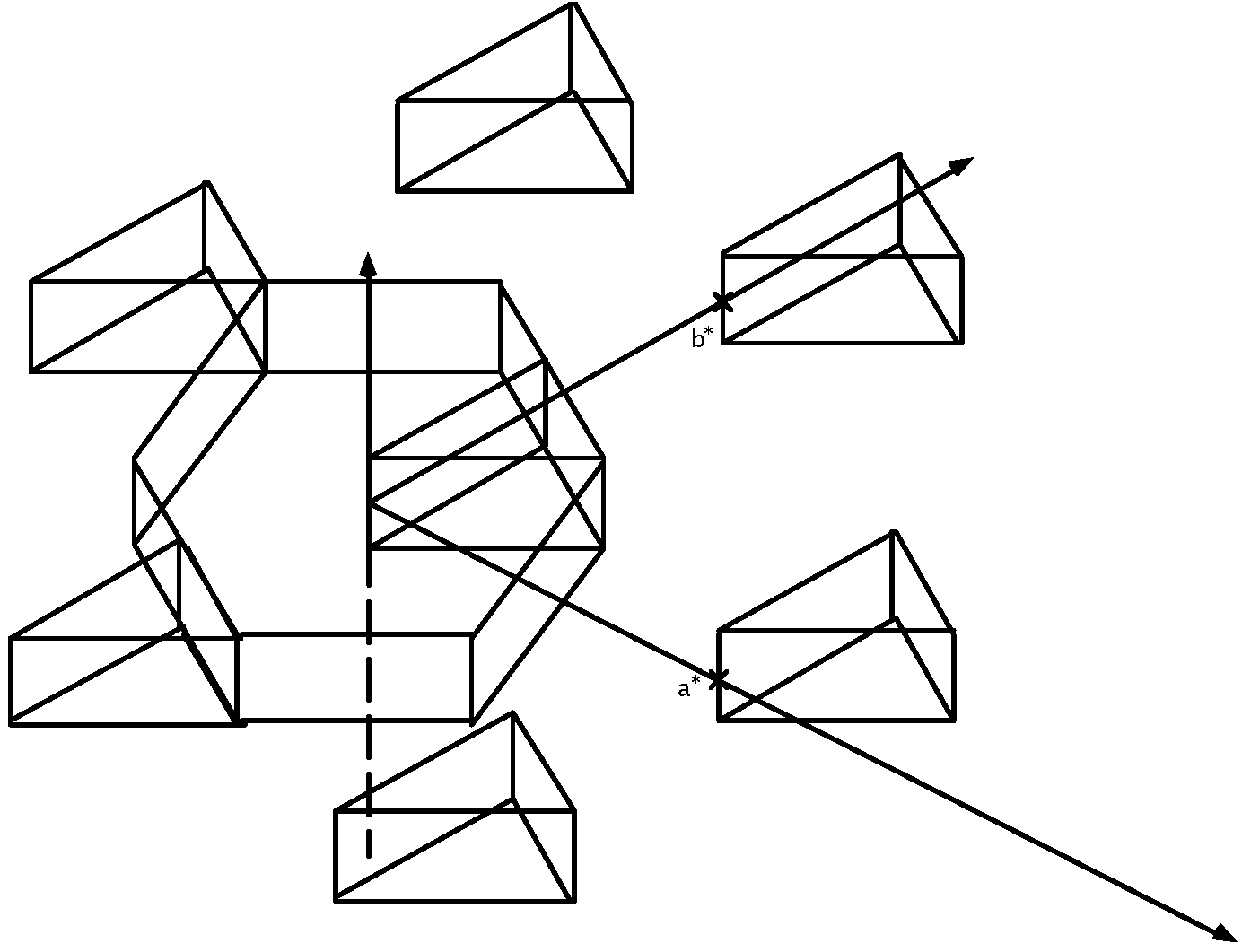
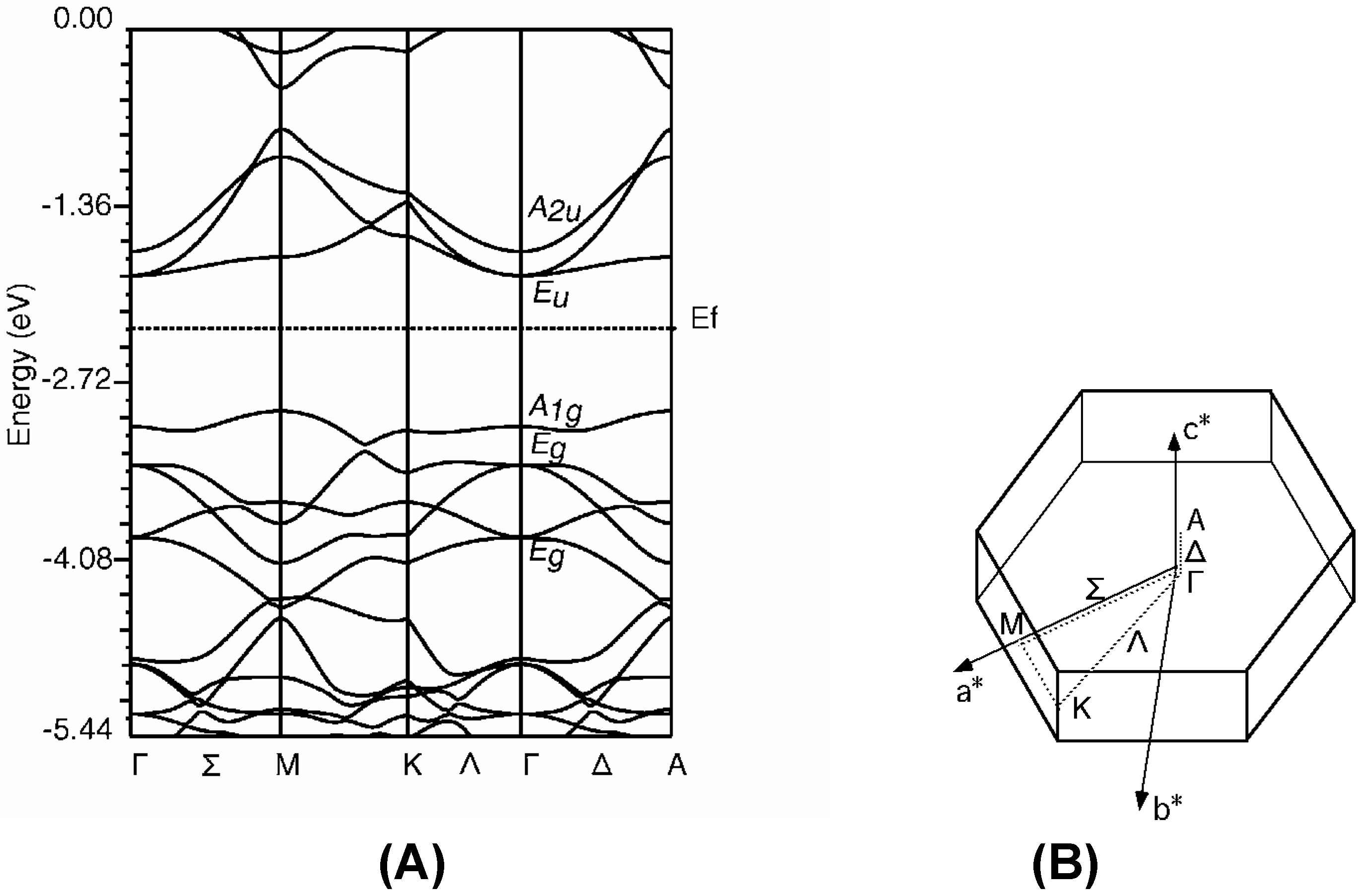
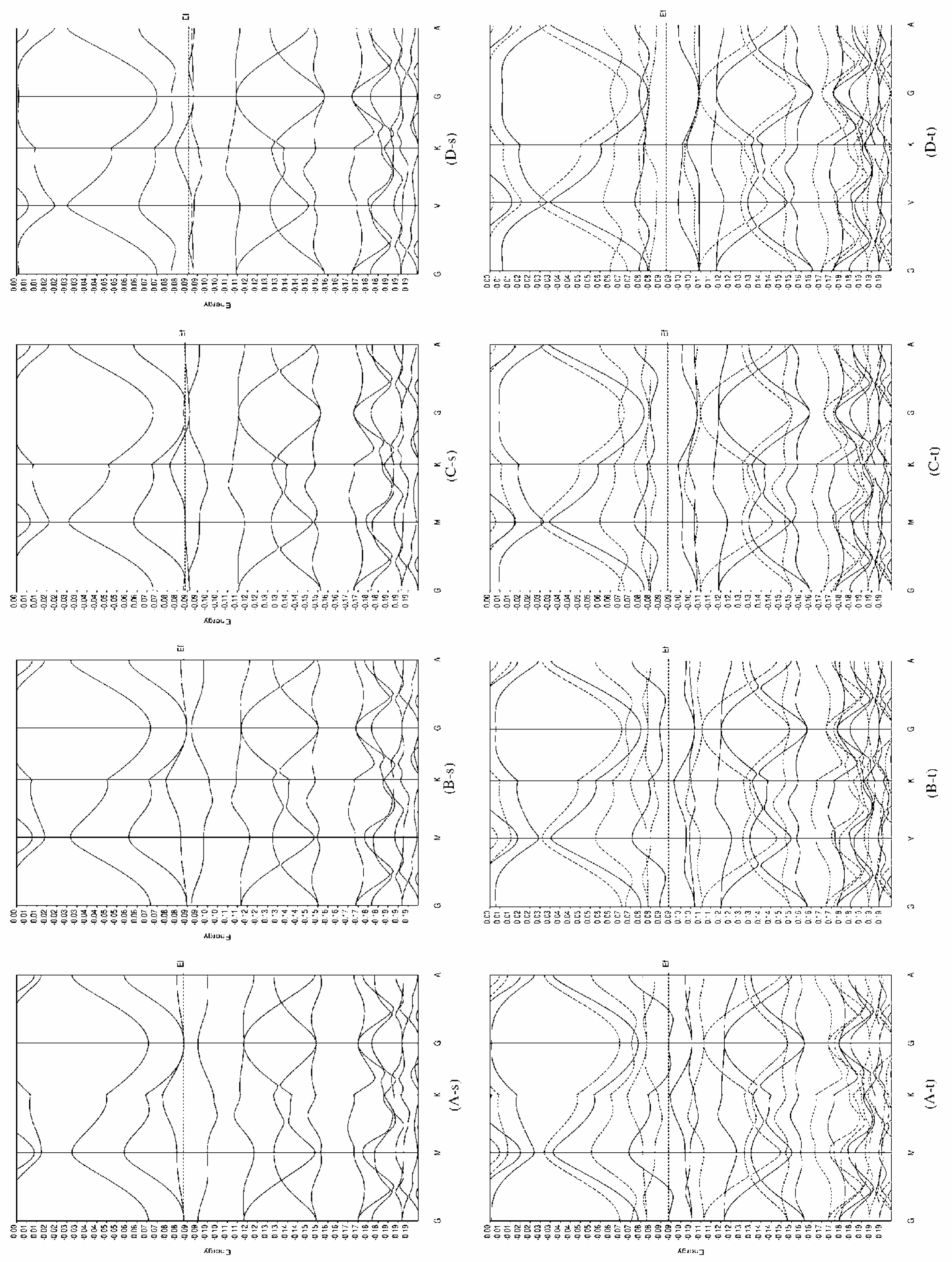
The band structure of Rh-C60
Group theoretical prediction








| Γ | E | 2C3 | 3C2 | i | 2S6 | 3σd |
|---|---|---|---|---|---|---|
| Γ1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Γ2 | 1 | 1 | -1 | 1 | 1 | -1 |
| Γ3 | 2 | -1 | 0 | 2 | -1 | 0 |
| Γ4 | 1 | 1 | 1 | -1 | -1 | -1 |
| Γ5 | 1 | 1 | -1 | -1 | -1 | 1 |
| Γ6 | 2 | -1 | 0 | -2 | 1 | 0 |









Rh-C60 without distortion of C60 cage
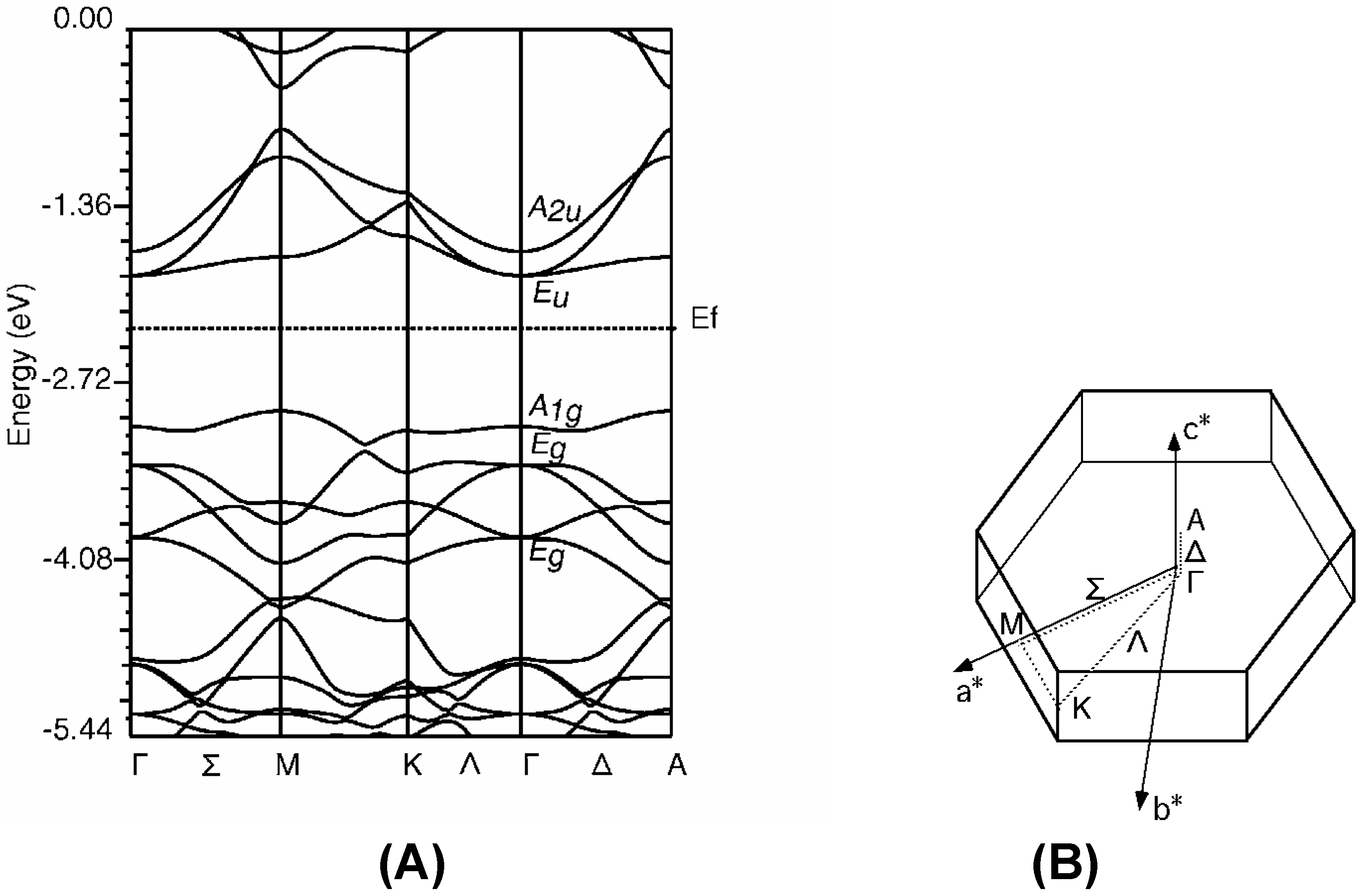
Rh-C60 with distortion of C60 cage

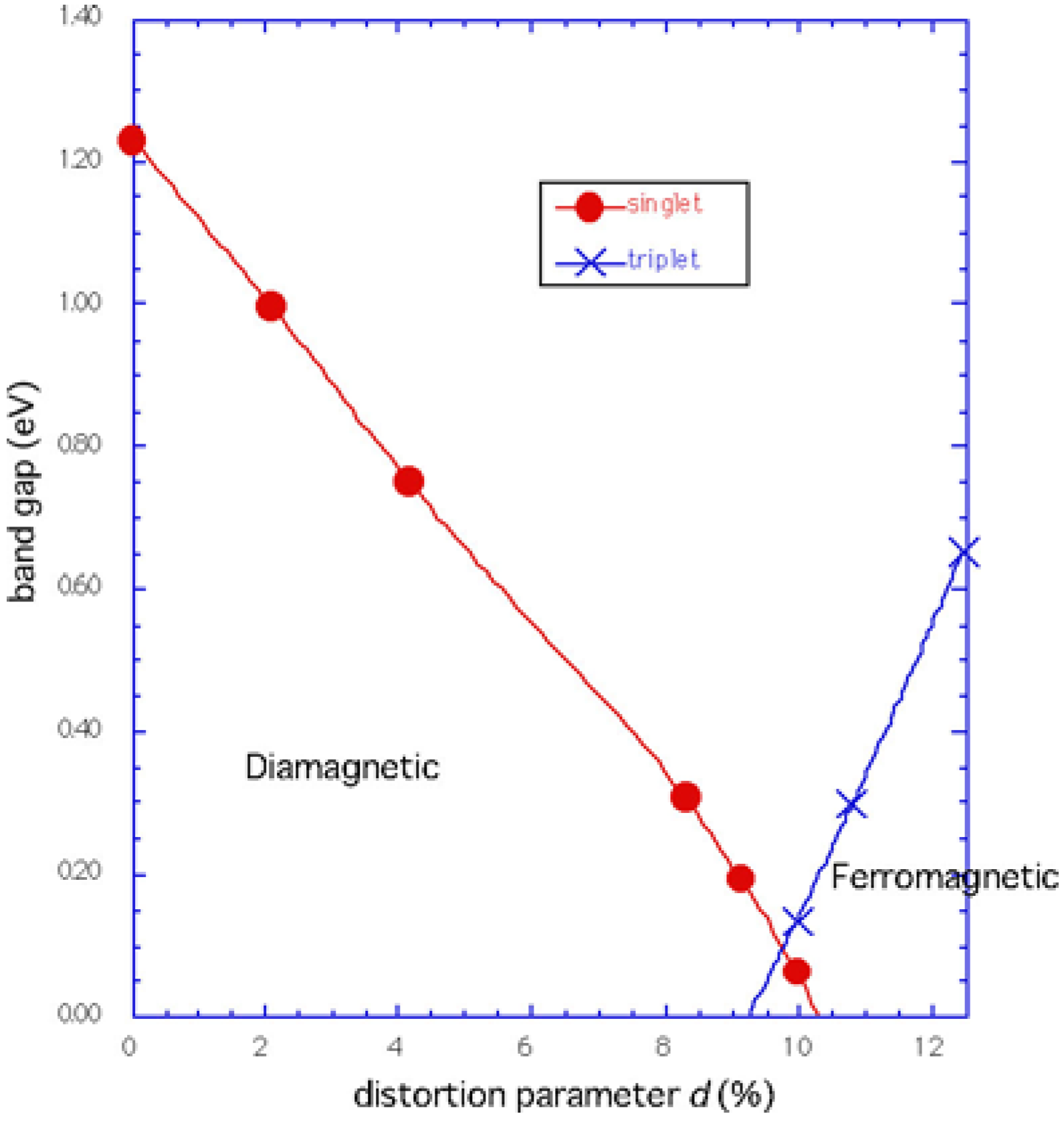
| d | Eg | EF |
|---|---|---|
| 0.00 | 1.23 | -2.30 |
| 2.08 | 1.00 | -2.30 |
| 4.15 | 0.75 | -2.30 |
| 8.30 | 0.31 | -2.24 |

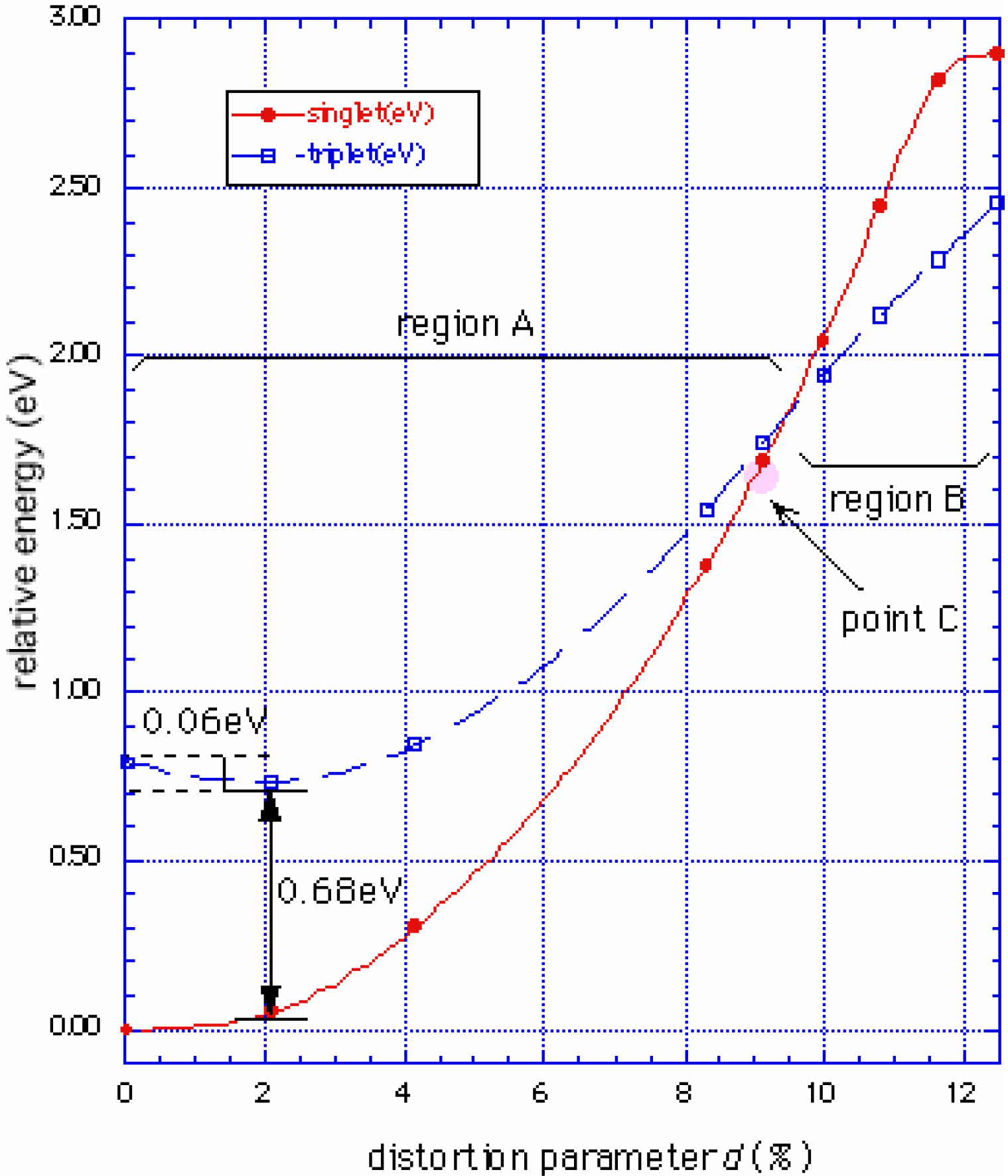


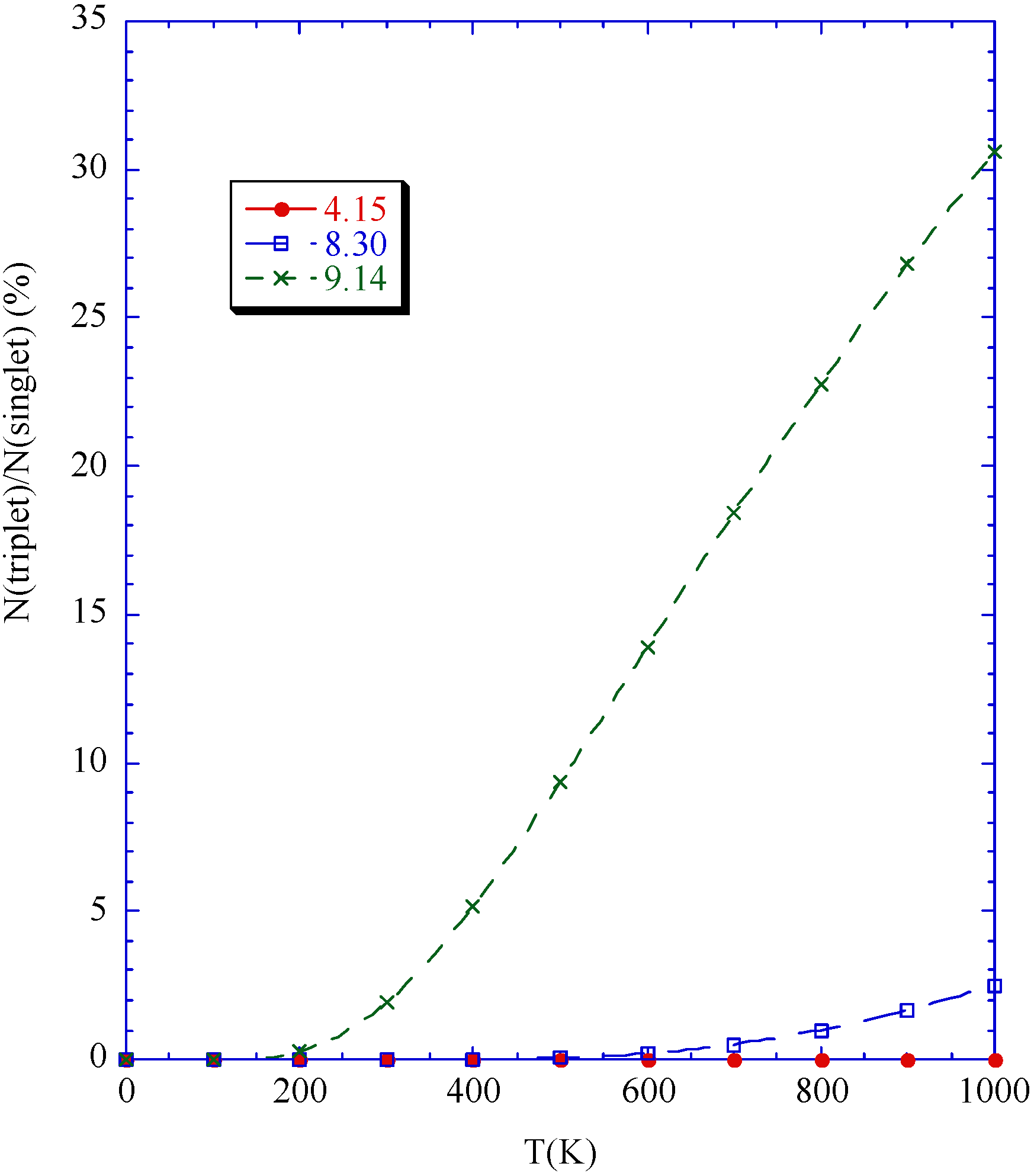
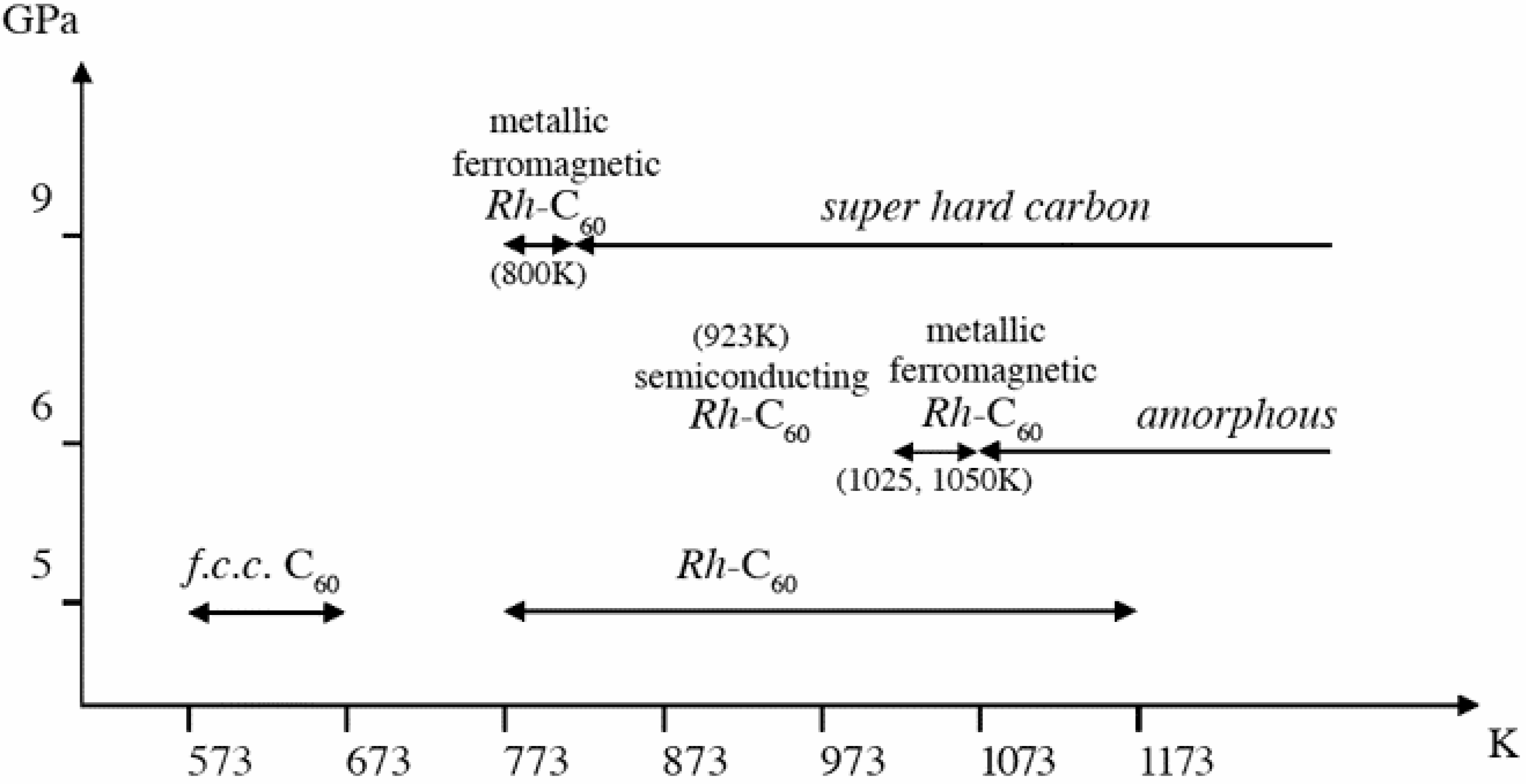
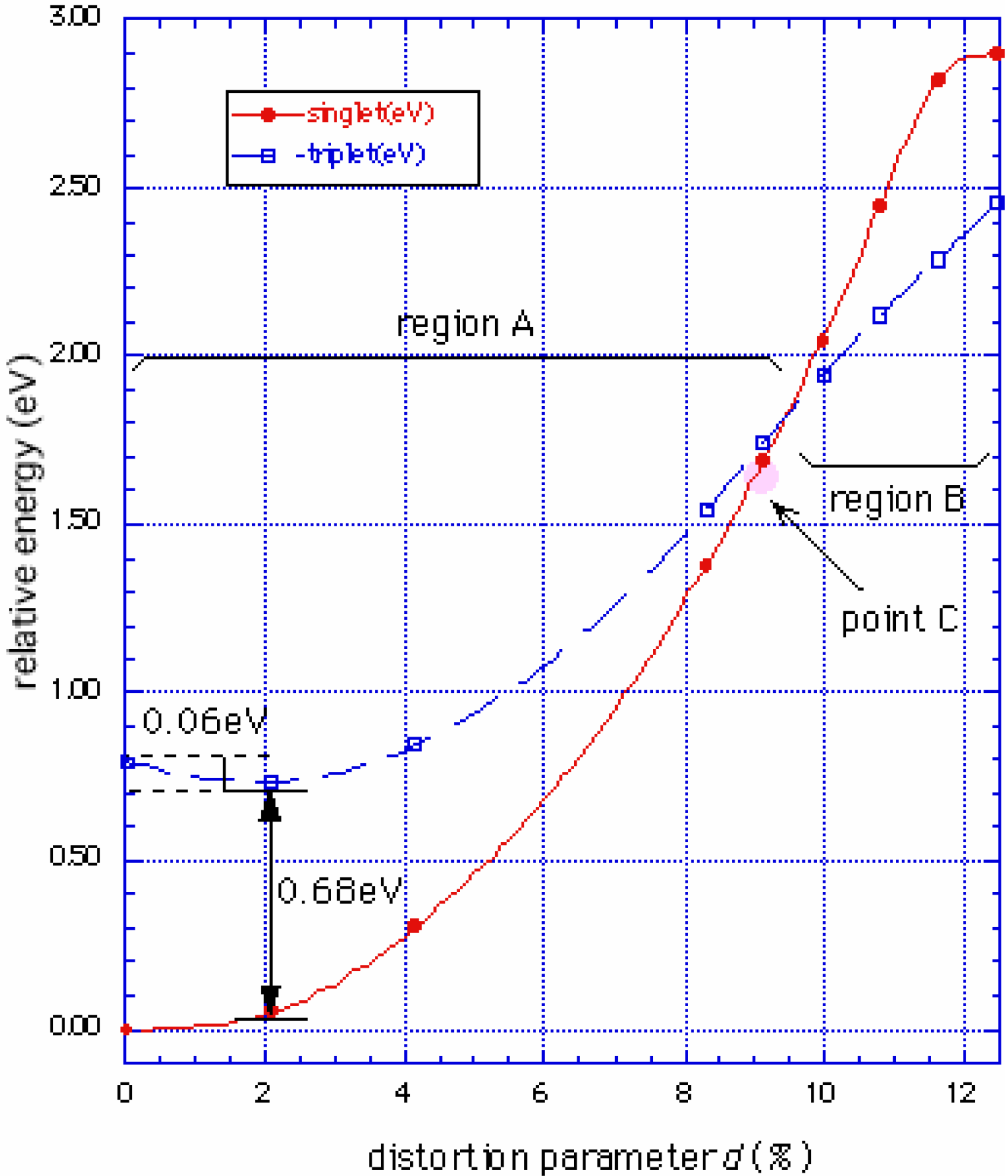
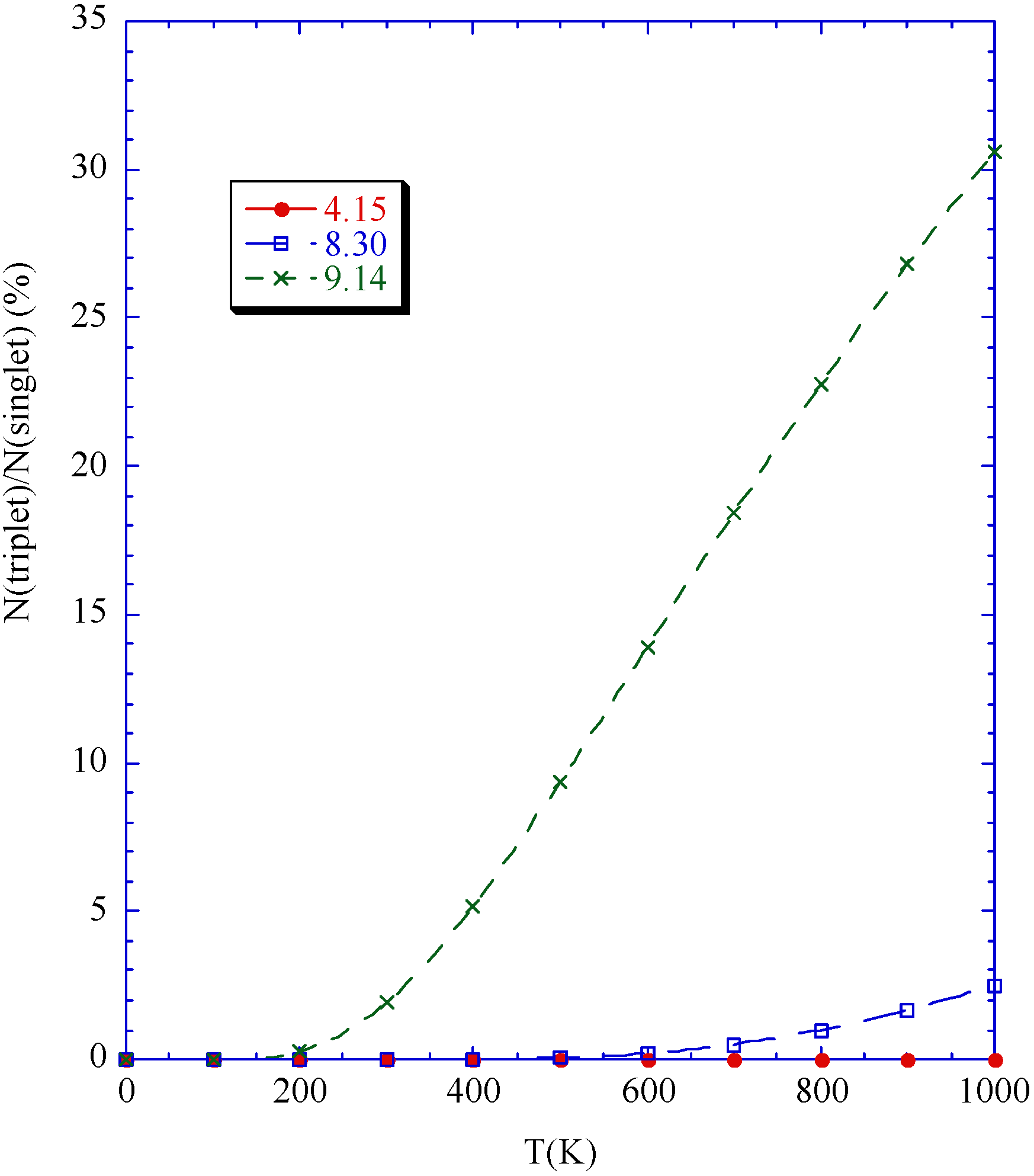
Phase diagram
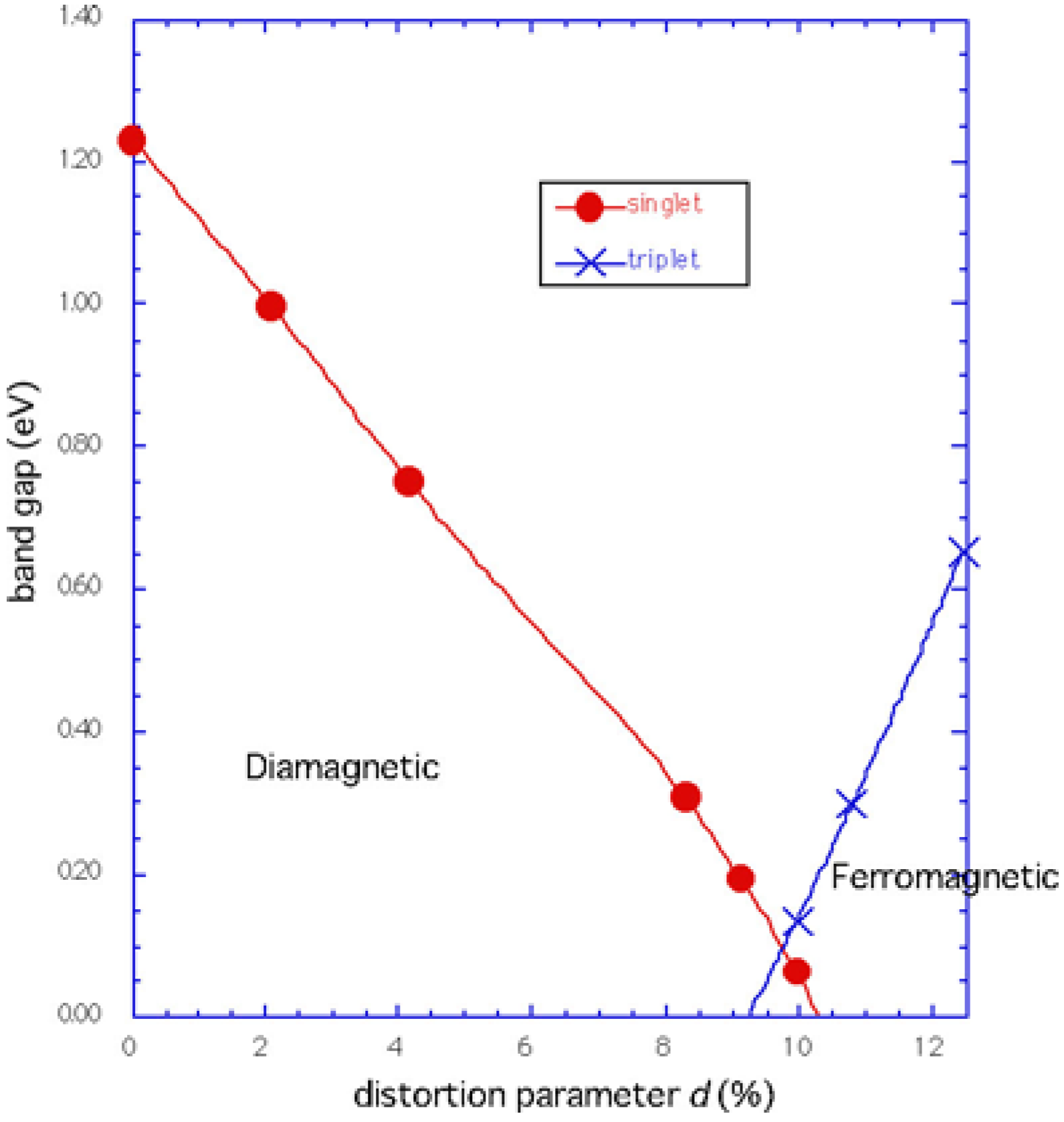
Conclusions
Supplementary Information
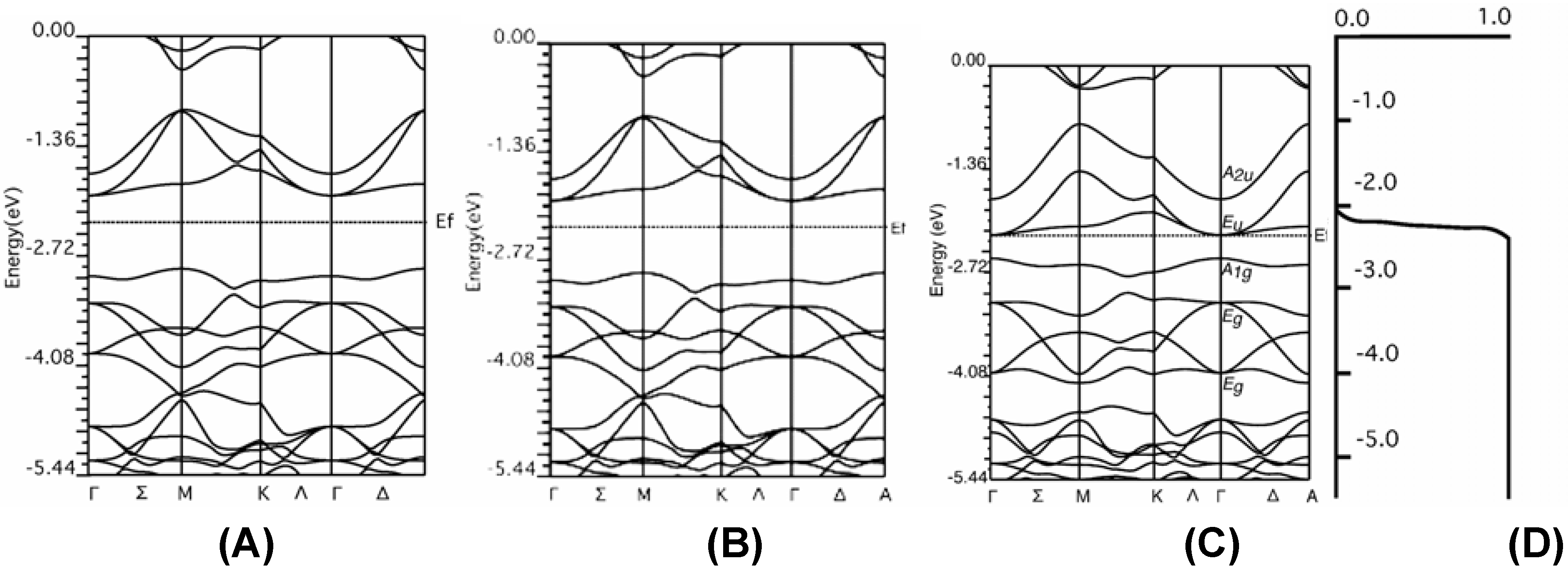
The verification of hybrid DFT band calculation
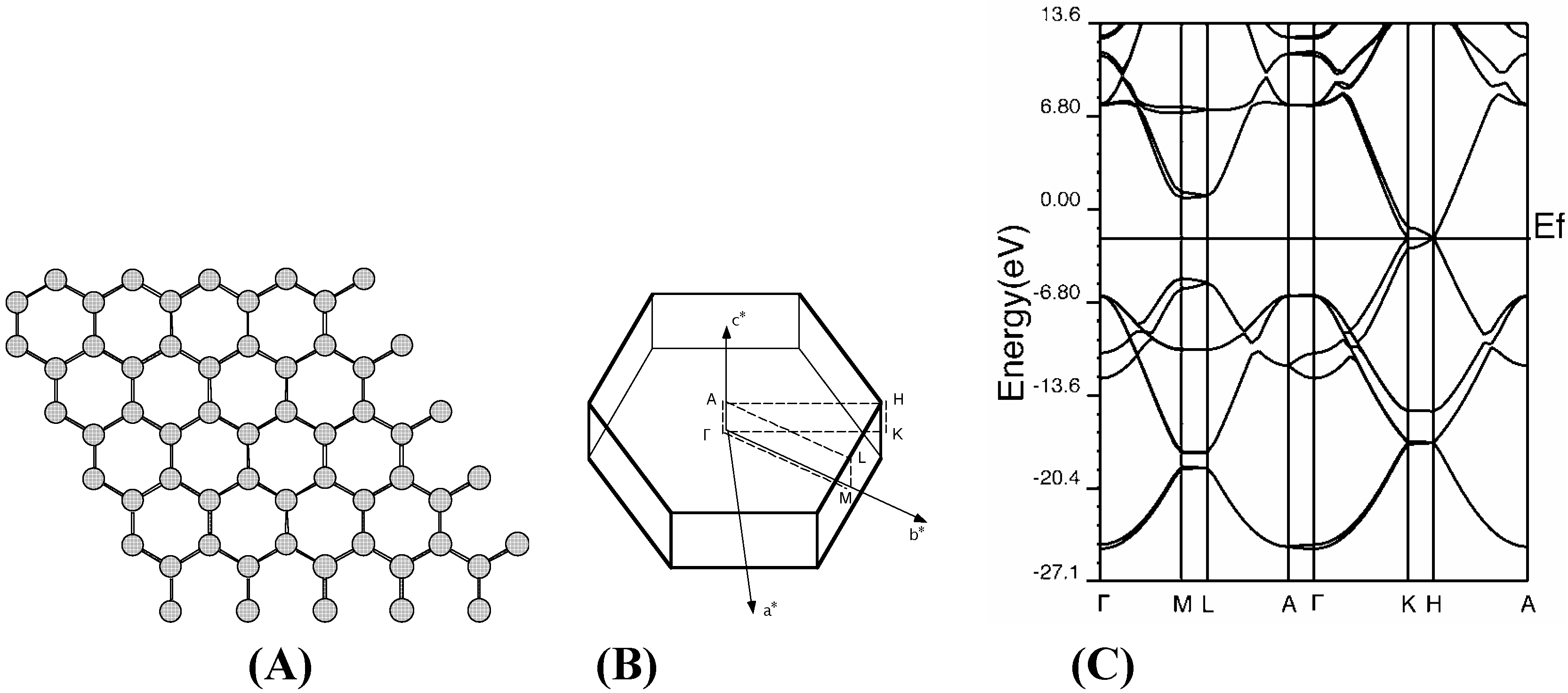
Graphite
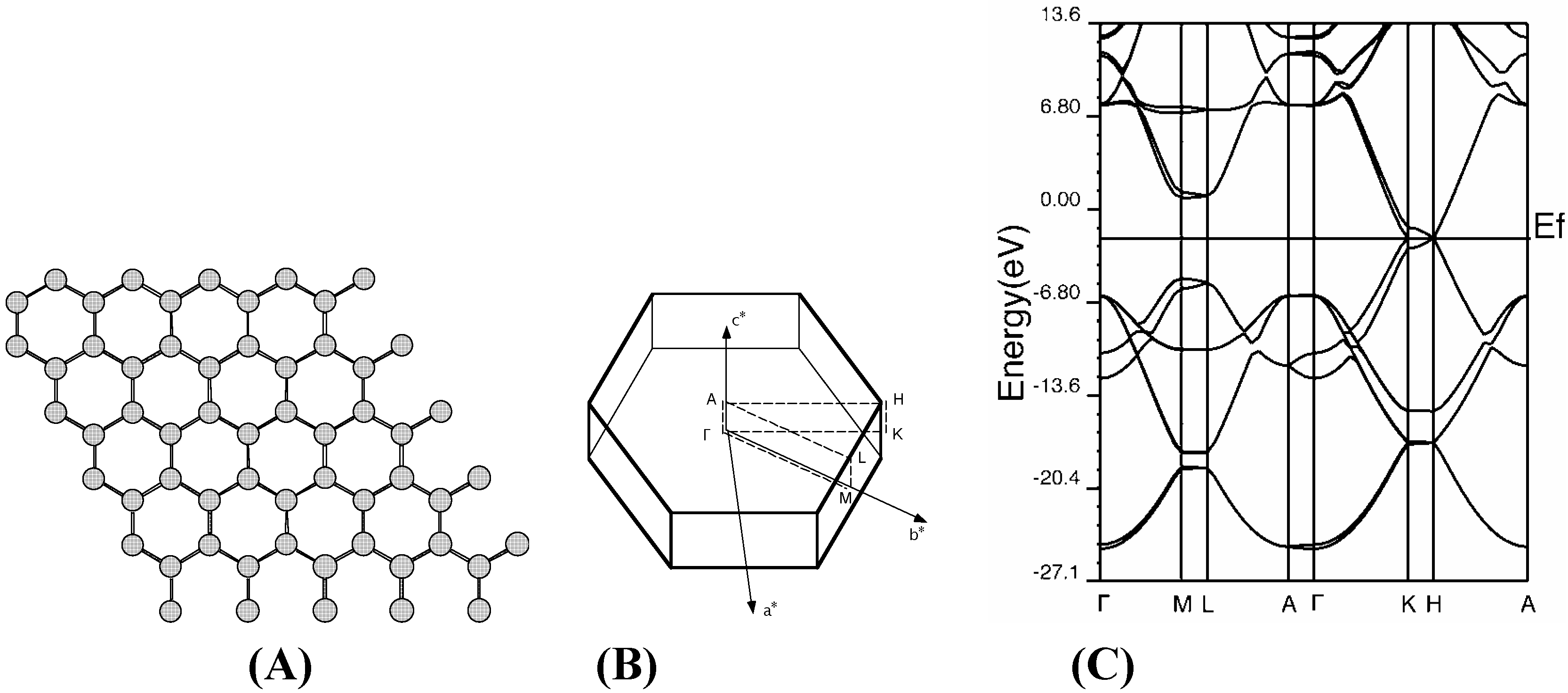
Diamond
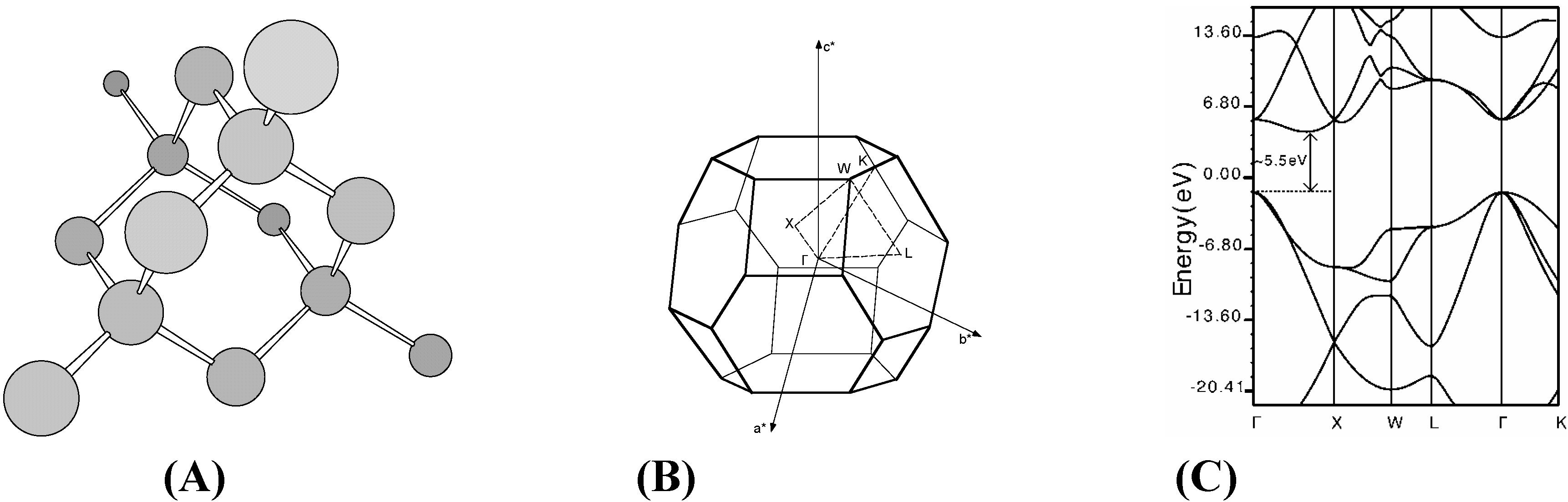
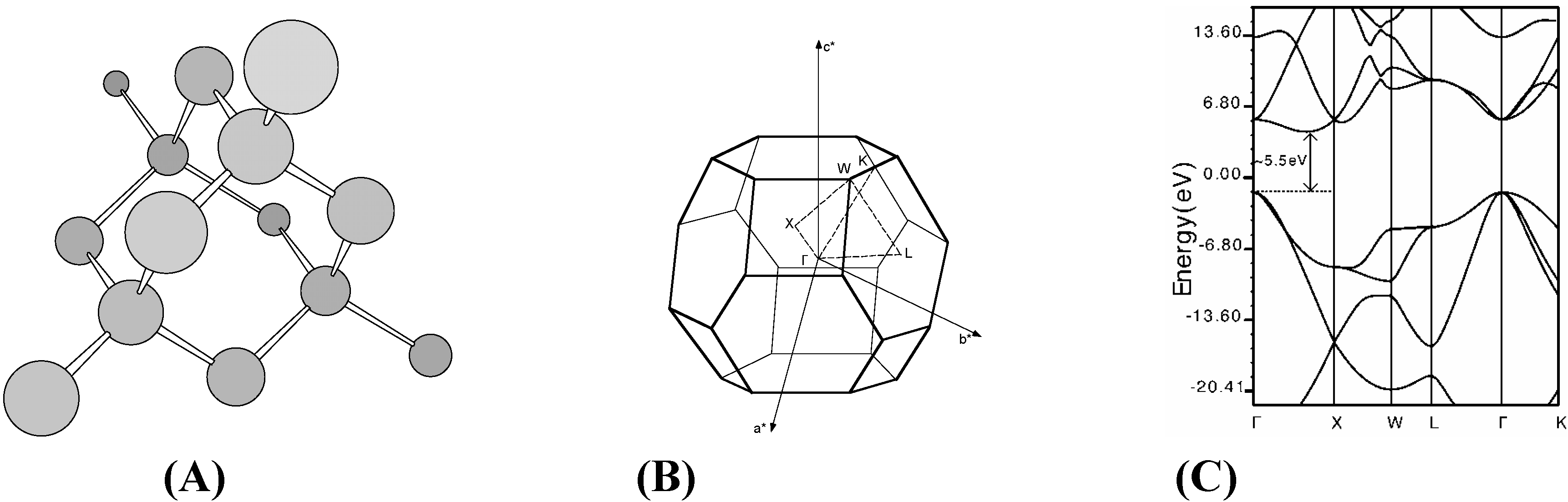
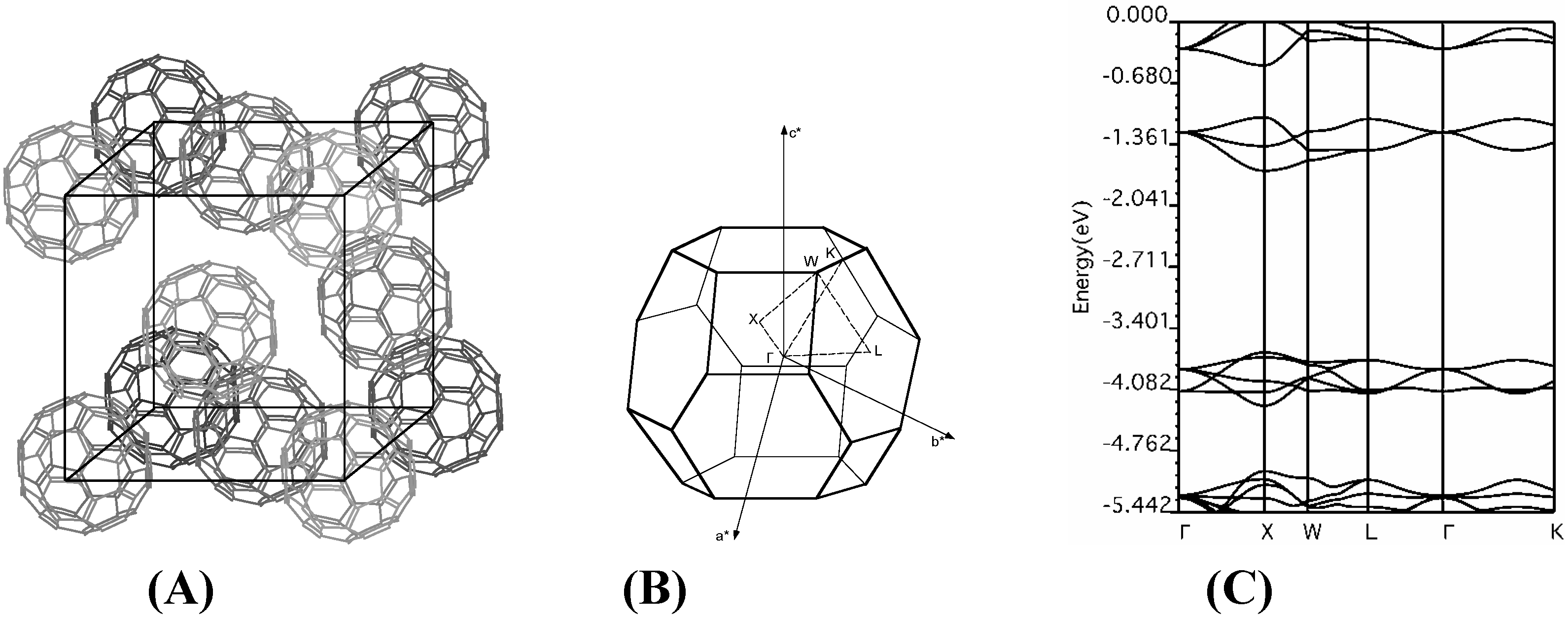
f.c.c. fullerene
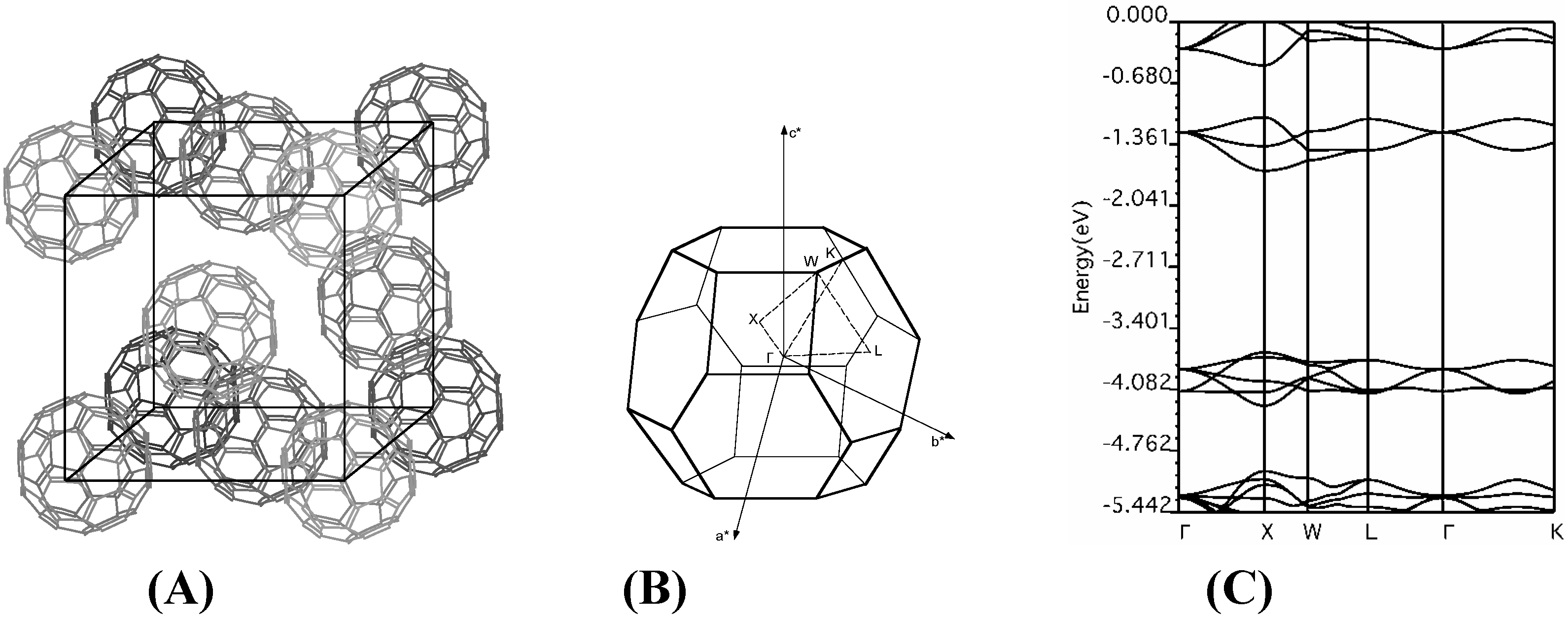
Acknowledgments
References and Notes
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. Nature 1985, 318, 162.
- Krätschmer, W.; Lamb, L.D.; Fostiropoulos, K.; Huffman, D.R. Nature 1990, 347, 354.
- Saito, S.; Oshiyama, A. Phys. Rev. Lett. 1991, 66, 2367.
- Haddon, R. C. Acc. Chem. Res. 1992, 25, 127.
- Iwasa, Y.; Arima, T.; Fleming, R.M.; Siegrist, T.; Zou, O.; Haddon, R.C.; Rothberg, L.J.; Lyons, K.B.; Carter Jr., H.L.; Hebard, A.F.; Tycko, R.; Dabbagh, G.; Krajewski, J.J.; Thomas, G.A.; Yagi, T. Science 1994, 264, 1570.
- Long, V.C.; Musfeldt, J.L.; Kamarás, K.; Adams, G.B.; Page, J.B.; Iwasa, Y.; Mayo, W.E. Phys Rev. 2001, B61, 13191.
- Davydov, V.A.; Kashevarova, L.S.; Rakhmanina, A.V.; Senyavin, V.M.; Céolin, R.; Szwarc, H.; Allouchi, H.; Agafonov, V. Phys Rev. 2000, B61, 11936.
- Makarova, T.M.; Sundqvist, B.; Höhne, R.; Esquinazi, P.; Kopelevich, Y.; Scharff, P.; Davydov, V.A.; Kashevarova, L.S.; Rakhmanina, A.V. Nature 2001, 413, 716.
- Allemand, P.-M.; Khemani, K.C.; Koch, A.; Wudl, F.; Holczer, K.; Donovan, S.; Grüner, G.; Thompson, J.D. Science 1991, 253, 301.
- Johnson, R. D.; Vries, M. S; Salem, J.; Bethune, D. S; Yannoni, C. S. Nature 1992, 355, 239.
- Tou, H.; Maniwa, Y.; Iwasa, Y.; Shimoda, H.; Mitani, T. Phys. Rev. 2000, B62, R775.
- Kitano, H.; Matsuo, R.; Miwa, K.; Maeda, A.; Takenobu, T.; Iwasa, Y.; Mitani, T. Phys. Rev. Lett. 2002, 88, 096401.
- Makarova, T.M.; Sundqvist, B.; Scharff, P.; Gaevski, M.E.; Olsson, E.; Davydov, V.A.; Rakhmanina, A.V.; Kashevarova, L.S. Carbon 2001, 39, 2203.
- Wood, R.A.; Lewis, M.H.; Lees, M.R.; Bennington, S.M.; Cain, M.G.; Kitamura, N. J. Phys. Condens. Matter 2002, 14, L385.
- It was written in many textbooks, for example: Kittel, C. Introduction to Solid State Physics; John Wiley & Sons, Inc.: New York, 1986. [Google Scholar].
- Erwin, S.C. Buckminsterfullerenes; Billups, W.E., Ciufolini, M.A., Eds.; VCH: New York, 1993; p. 217. [Google Scholar]
- Mielke, A.; Tasaki, H. Commun. Math. Phys. 1993, 158, 341.
- For instance, ѱ(Γ3)i was obtained as follows;
where l and h denote the dimension and order of symmetry on consideration.
![Molecules 09 00792 i020]()
- Han, K.-H.; Spemann, D.; Höhne, R.; Setzet, A.; Makarova, T.; Esquinazi, P.; Butz, T. Carbon 2003, 41, 785.
- Andriotis, A. N.; Menon, M.; Sheetz, R. M.; Chernozatonskii, L. Phys. Rev. Lett. 2003, 90, 026801.
- Ribas-Ariño, J.; Novoa, J. J. J. Phys. Chem. Solids. 2004, 65, 787.
- Okada, S.; Oshiyama, A. Phys. Rev. 2003, B68, 23542.
- Chi, D. H.; Iwasa, Y.; Takano, T.; Watanuki, T.; Ohishi, Y.; Yamanaka, S. Phys. Rev. 2003, B68, 153402.
- Korobov, M. V.; Senyavin, V. M; Bogachev, A. G.; Stukalin, E. B.; Davidov, V. A.; Kashevarona, L. S.; Rakhmanina, A. V.; Agafonov, V.; Szwarc, A. Chem. Phys. Lett. 2003, 381, 410.
- Makarova, T. L.; Han, K.-H.; Esquinazi, P.; da Silva, R. R.; Kopelevich, Y.; Zakharova, I. B.; Sundqvist, B. Carbon 2003, 41, 1575.
- Owens, F. J.; Iqhal, Z.; Belova, L.; Rao, K. V. Phys. Rev. 2004, B69, 033403.
- Nakano, S.; Kitagawa, Y.; Kawakami, T.; Yamaguchi, T. Synthetic Metals 2003, 135-136, 779.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Samples Availability: Available from the authors.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Nakano, S.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Nagao, H.; Yamaguchi, K. Theoretical Studies on Electronic States of Rh-C60. Possibility of a Room-temperature Organic Ferromagnet. Molecules 2004, 9, 792-807. https://doi.org/10.3390/90900792
Nakano S, Kitagawa Y, Kawakami T, Okumura M, Nagao H, Yamaguchi K. Theoretical Studies on Electronic States of Rh-C60. Possibility of a Room-temperature Organic Ferromagnet. Molecules. 2004; 9(9):792-807. https://doi.org/10.3390/90900792
Chicago/Turabian StyleNakano, S., Y. Kitagawa, T. Kawakami, M. Okumura, H. Nagao, and K. Yamaguchi. 2004. "Theoretical Studies on Electronic States of Rh-C60. Possibility of a Room-temperature Organic Ferromagnet" Molecules 9, no. 9: 792-807. https://doi.org/10.3390/90900792