Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c

Abstract
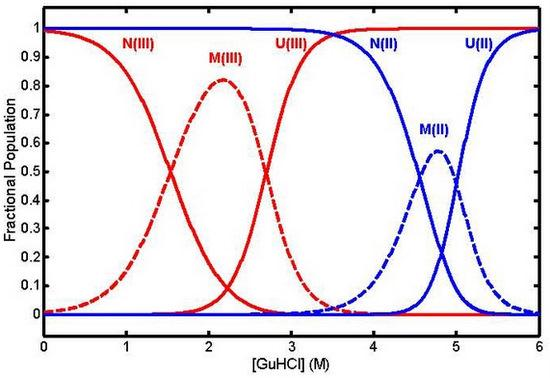
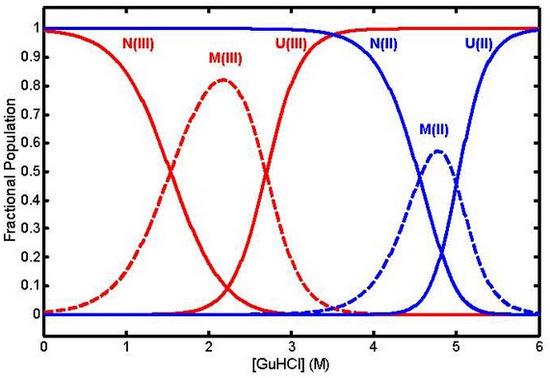
:
{kind=link}
{kind=link}
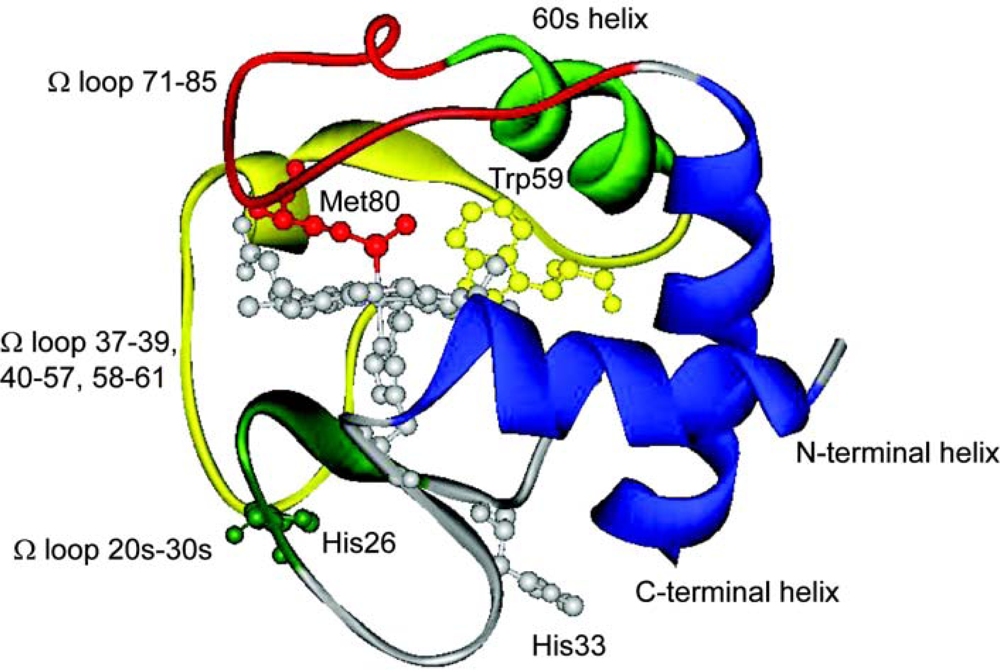{kind=link}
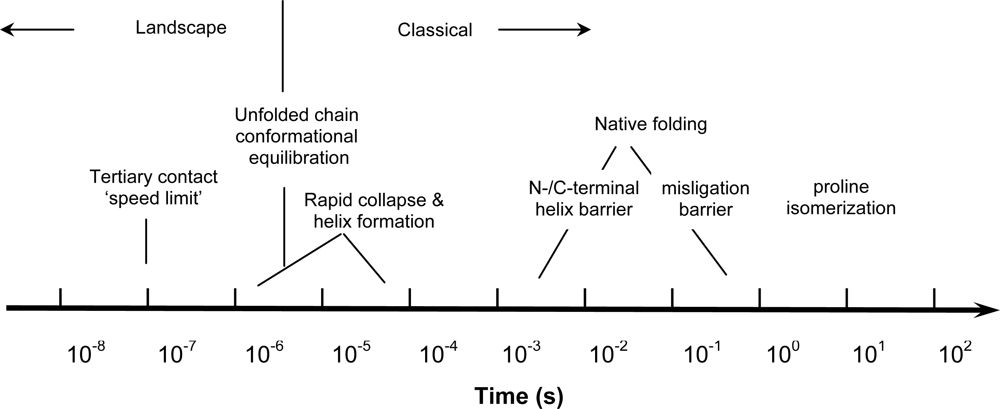{kind=link}
{kind=link}
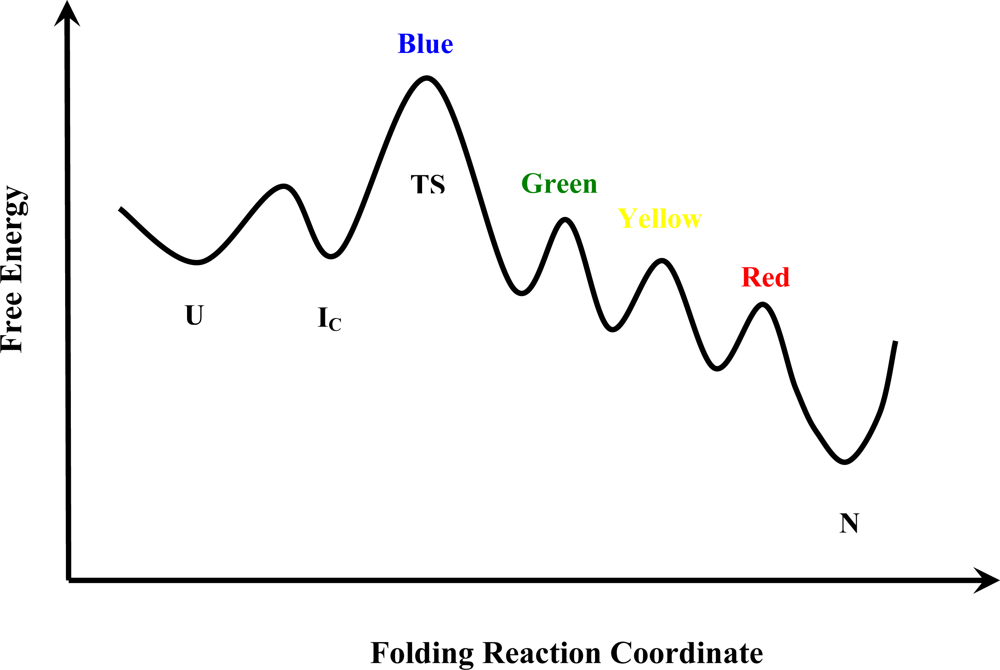{kind=link}
1. Introduction
2. Late Folding Processes (t > 1 ms)
3. The Nature of the Unfolded State
4. The Dynamics of the Unfolded State
5. Earliest Folding Events (t < 1 ms)
5.1. Kinetic Evidence For a Free Energy Barrier to Rapid Collapse
5.2. Submillisecond Secondary Structure Formation
5.3. Are Rapid Collapse and Helix Formation Simultaneous?
6. Molten Globules as Models for Folding Intermediates
7. Is the Microsecond-Timescale Collapsed Intermediate (IC) a Sequence-Specific Folding Intermediate?
8. Is IC a Molten Globule?
9. Is IC on Pathway?
10. Implications for Folding Mechanisms
11. Other Cytochromes c
12. Conclusions
References
- Jones, CM; Henry, ER; Hu, Y; Chan, CK; Luck, SD; Bhuyan, A; Roder, H; Hofrichter, J; Eaton, WA. Fast events in protein folding initiated by nanosecond laser photolysis. Proc. Natl. Acad. Sci. USA 1993, 90, 11860–11864. [Google Scholar]
- Pascher, T; Chesick, JP; Winkler, JR; Gray, HB. Protein folding triggered by electron transfer. Science 1996, 271, 1556–1560. [Google Scholar]
- Chan, CK; Hofrichter, J; Eaton, WA; Winkler, JR; Gray, HB. Optical triggers of protein folding. Science 1996, 274, 628–629. [Google Scholar]
- Babul, J; Stellwagen, E. The existence of heme-protein coordinate-covalent bonds in denaturing solvents. Biopolymers 1971, 10, 2359–2361. [Google Scholar]
- Muthukrishnan, K; Nall, BT. Effective concentrations of amino acid side chains in an unfolded protein. Biochemistry 1991, 30, 4706–4710. [Google Scholar]
- Chen, E; Wood, MJ; Fink, AL; Kliger, DS. Time-resolved circular dichroism studies of protein folding intermediates of cytochrome c. Biochemistry 1998, 37, 5589–5598. [Google Scholar]
- Brems, DN; Stellwagen, E. Manipulation of the observed kinetic phases in the refolding of denatured ferricytochrome c. J. Biol. Chem 1983, 258, 3655–3660. [Google Scholar]
- Winkler, JR. Cytochrome c folding dynamics. Curr. Opin. Chem. Biol 2004, 8, 169–174. [Google Scholar]
- Sosnick, TR; Mayne, L; Hiller, R; Englander, SW. The barriers in protein folding. Nat. Struct. Biol 1994, 1, 149–156. [Google Scholar]
- Roder, H; Elöve, GA; Englander, SW. Structural characterization of folding intermediates in cytochrome c by H-exchange labelling and proton NMR. Nature 1988, 335, 700–704. [Google Scholar]
- Krishna, MMG; Lin, Y; Mayne, L; Englander, SW. Intimate view of a kinetic protein folding intermediate: Residue-resolved structure, interactions, stability, folding and unfolding rates, homogeneity. J. Mol. Biol 2003, 334, 501–513. [Google Scholar]
- Krishna, MMG; Lin, Y; Englander, SW. Protein misfolding: Optional barriers, misfolded intermediates, and pathway heterogeneity. J. Mol. Biol 2004, 343, 1095–1109. [Google Scholar]
- Sosnick, TR; Mayne, L; Englander, SW. Molecular collapse: the rate-limiting step in two-state cytochrome c folding. Proteins 1996, 24, 413–426. [Google Scholar]
- Mines, GA; Pascher, T; Lee, SC; Winkler, JR; Gray, HB. Cytochrome c folding triggered by electron transfer. Chem. Biol 1996, 3, 491–497. [Google Scholar]
- Hammond, GS. A correlation of reaction rates. J. Am. Chem. Soc 1955, 77, 334–338. [Google Scholar]
- Bai, YW; Sosnick, TR; Mayne, L; Englander, SW. Protein folding intermediates: Native-state hydrogen exchange. Science 1995, 269, 192–197. [Google Scholar]
- Krishna, MMG; Lin, Y; Rumbley, JN; Englander, SW. Cooperative omega loops in cytochrome c: Role in folding and function. J. Mol. Biol 2003, 331, 29–36. [Google Scholar]
- Englander, SW; Mayne, L; Rumbley, JN. Submolecular cooperativity produces multi-state protein unfolding and refolding. Biophys Chem 2002, 101–102, 57–65. [Google Scholar]
- Krantz, BA; Mayne, L; Rumbley, J; Englander, SW; Sosnick, TR. Fast and slow intermediate accumulation and the initial barrier mechanism in protein folding. J. Mol. Biol 2002, 324, 359–371. [Google Scholar]
- Pletneva, EV; Gray, HB; Winkler, JR. Many faces of the unfolded state: Conformational heterogeneity in denatured yeast cytochrome c. J. Mol. Biol 2005, 345, 855–867. [Google Scholar]
- Segel, DJ; Fink, AL; Hodgson, KO; Doniach, S. Protein denaturation: A small-angle X-ray scattering study of the ensemble of unfolded states of cytochrome c. Biochemistry 1998, 37, 12443–12451. [Google Scholar]
- Sauder, JM; Roder, H. Amide protection in an early folding intermediate of cytochrome c. Fold. Des 1998, 3, 293–301. [Google Scholar]
- Colon, W; Wakem, LP; Sherman, F; Roder, H. Identification of the predominant non-native histidine ligand in unfolded cytochrome c. Biochemistry 1997, 36, 12535–12541. [Google Scholar]
- Thomas, YG; Goldbeck, RA; Kliger, DS. Characterization of equilibrium intermediates in denaturant-induced unfolding of ferrous and ferric cytochromes c using magnetic circular dichroism, circular dichroism, and optical absorption spectroscopies. Biopolymers 2000, 57, 29–36. [Google Scholar]
- Hagen, SJ; Hofrichter, J; Szabo, A; Eaton, WA. Diffusion-limited contact formation in unfolded cytochrome c: Estimating the maximum rate of protein folding. Proc. Natl. Acad. Sci. USA 1996, 93, 11615–11617. [Google Scholar]
- Camacho, CJ; Thirumalai, D. Theoretical predictions of folding pathways using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA 1995, 92, 1277–1281. [Google Scholar]
- Goldbeck, RA; Thomas, YG; Chen, E; Esquerra, RM; Kliger, DS. Multiple pathways on a protein-folding energy landscape: Kinetic evidence. Proc. Natl. Acad. Sci. USA 1999, 96, 2782–2787. [Google Scholar]
- Chen, E; Goldbeck, RA; Kliger, DS. Earliest events in protein folding: Submicrosecond secondary structure formation in reduced cytochrome c. J. Phys. Chem. A 2003, 107, 8149–8155. [Google Scholar]
- Chang, IJ; Lee, JC; Winkler, JR; Gray, HB. The protein-folding speed limit: Intrachain diffusion times set by electron-transfer rates in denatured Ru(NH3)5(His-33)-Zn-cytochrome c. Proc. Natl. Acad. Sci. USA 2003, 100, 3838–3840. [Google Scholar]
- Abel, CJ; Goldbeck, RA; Latypov, RF; Roder, H; Kliger, DS. Conformational equilibration time of unfolded protein chains and the folding speed limit. Biochemistry 2007, 46, 4090–4099. [Google Scholar]
- Bieri, O; Wirz, J; Hellrung, B; Schutkowski, M; Drewello, M; Kiefhaber, T. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc. Natl. Acad. Sci. USA 1999, 96, 9597–9601. [Google Scholar]
- Lapidus, LJ; Eaton, WA; Hofrichter, J. Measuring the rate of intramolecular contact formation in polypeptides. Proc. Natl. Acad. Sci. USA 2000, 97, 7220–7225. [Google Scholar]
- Krieger, F; Fierz, B; Bieri, O; Drewello, M; Kiefhaber, T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J. Mol. Biol 2003, 332, 265–274. [Google Scholar]
- Yang, WY; Gruebele, M. Folding at the speed limit. Nature 2003, 423, 193–197. [Google Scholar]
- Socci, ND; Onuchic, JN; Wolynes, PG. Diffusive dynamics of the reaction coordinate for protein folding funnels. J. Chem. Phys 1996, 104, 5860–5868. [Google Scholar]
- Portman, JJ; Takada, S; Wolynes, PG. Microscopic theory of protein folding rates. II. Local reaction coordinates and chain dynamics. J. Chem. Phys 2001, 114, 5082–5096. [Google Scholar]
- Pascher, T. Temperature and driving force dependence of the folding rate of reduced horse heart cytochrome c. Biochemistry 2001, 40, 5812–5820. [Google Scholar]
- Eaton, WA; Thompson, PA; Chan, CK; Hagen, SJ; Hofrichter, J. Fast events in protein folding. Structure 1996, 4, 1133–1139. [Google Scholar]
- Kubelka, J; Hofrichter, J; Eaton, WA. The protein folding ‘speed limit’. Curr. Opin. Struct. Biol 2004, 14, 76–88. [Google Scholar]
- Chan, CK; Hu, Y; Takahashi, S; Rousseau, DL; Eaton, WA; Hofrichter, J. Submillisecond protein folding kinetics studied by ultrarapid mixing. Proc. Natl. Acad. Sci. USA 1997, 94, 1779–1784. [Google Scholar]
- Shastry, MCR; Roder, H. Evidence for barrier-limited protein folding kinetics on the microsecond timescale. Nat. Struct. Biol 1998, 5, 385–392. [Google Scholar]
- Hagen, SJ; Eaton, WA. Two-state expansion and collapse of a polypeptide. J. Mol. Biol 2000, 297, 781–789. [Google Scholar]
- Qiu, L; Zachariah, C; Hagen, SJ. Fast chain contraction during protein folding: “Foldability” and collapse dynamics. Phys. Rev. Lett 2003, 90, 168103. [Google Scholar]
- Lyubovitsky, JG; Gray, HB; Winkler, JR. Mapping the cytochrome c folding landscape. J. Am. Chem. Soc 2002, 124, 5481–5485. [Google Scholar]
- Pletneva, EV; Gray, HB; Winkler, JR. Snapshots of cytochrome c folding. Proc. Natl. Acad. Sci. USA 2005, 102, 18397–18402. [Google Scholar]
- Sosnick, TR; Shtilerman, MD; Mayne, L; Englander, SW. Ultrafast signals in protein folding and the polypeptide contracted state. Proc. Natl. Acad. Sci. USA 1997, 94, 8545–8550. [Google Scholar]
- Ptitsyn, OB. Stages in the mechanism of self-organization of protein molecules. Dokl. Akad. Nauk SSSR 1973, 210, 1213–1215. [Google Scholar]
- Kim, PS; Baldwin, RL. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Ann. Rev. Biochem 1982, 51, 459–489. [Google Scholar]
- Ptitsyn, OB. Molten globule and protein folding. Adv. Protein Chem 1995, 47, 83–229. [Google Scholar]
- Dagget, V; Fersht, AR. Is there a unifying mechanism for protein folding? Trends Biochem. Sci 2003, 28, 18–25. [Google Scholar]
- Pollack, L; Tate, MW; Darnton, NC; Knight, JB; Gruner, SM; Eaton, WA; Austin, RH. Compactness of the denatured state of a fast-folding protein measured by submillisecond smallangle x-ray scattering. Proc. Natl. Acad. Sci. USA 1999, 96, 10115–10117. [Google Scholar]
- Lee, JC; Chang, IJ; Gray, HB; Winkler, JR. The cytochrome c folding landscape revealed by electron-transfer kinetics. J. Mol. Biol 2002, 320, 159–164. [Google Scholar]
- Tezcan, FA; Findley, WM; Crane, BR; Ross, SA; Lyubovitsky, JG; Gray, HB; Winkler, JR. Using deeply trapped intermediates to map the cytochrome c folding landscape. Proc. Natl. Acad. Sci. USA 2002, 99, 8626–8630. [Google Scholar]
- Pletneva, EV; Gray, HB; Winkler, JR. Nature of the cytochrome c molten globule. J. Am. Chem. Soc 2005, 127, 15370–15371. [Google Scholar]
- Elove, GA; Chaffotte, AF; Roder, H; Goldberg, ME. Early steps in cytochrome c folding probed by time-resolved circular dichroism and fluorescence spectroscopy. Biochemistry 1992, 31, 6876–6883. [Google Scholar]
- Chen, E; Wittung-Stafshede, P; Kliger, DS. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc 1999, 121, 3811–3817. [Google Scholar]
- Chen, E; Goldbeck, RA; Kliger, DS. The earliest events in protein folding: A structural requirement for ultrafast folding in cytochrome c. J. Am. Chem. Soc 2004, 126, 11175–11181. [Google Scholar]
- Chen, E; Abel, CJ; Goldbeck, RA; Kliger, DS. Non-native heme–histidine ligation promotes microsecond time scale secondary structure formation in reduced horse heart cytochrome c. Biochemistry 2007, 46, 12463–12472. [Google Scholar]
- Akiyama, S; Takahashi, S; Ishimori, K; Morishima, I. Stepwise formation of α-helices during cytochrome c folding. Nat. Struct. Biol 2000, 7, 514–520. [Google Scholar]
- Uversky, VN; Fink, AL. The chicken-egg scenario of protein folding revisited. FEBS Lett 2002, 515, 79–83. [Google Scholar]
- Jennings, PA; Wright, PE. Formation of a molten globule intermediate early in the kinetic folding pathway of apomyoglobin. Science 1993, 262, 892–896. [Google Scholar]
- Raschke, TM; Marqusee, S. The kinetic folding intermediate of ribonuclease H resembles the acid molten globule and partially unfolded molecules detected under native conditions. Nat. Struct. Biol 1997, 4, 298–304. [Google Scholar]
- Colon, W; Roder, H. Kinetic intermediates in the formation of the cytochrome c molten globule. Nat. Struct. Biol 1996, 3, 1019–1025. [Google Scholar]
- Pabit, SA; Roder, H; Hagen, SJ. Internal friction controls the speed of protein folding from a compact configuration. Biochemistry 2004, 43, 12532–12538. [Google Scholar]
- Latypov, RF; Maki, K; Cheng, H; Luck, SD; Roder, H. Folding mechanism of reduced cytochrome c: Equilibrium and kinetic properties in the presence of carbon monoxide. J. Mol. Biol 2008, 383, 437–453. [Google Scholar]
- Tezcan, FA; Winkler, JR; Gray, HB. Effects of ligation and folding on reduction potentials of heme proteins. J. Am. Chem. Soc 1998, 120, 13383–13388. [Google Scholar]
- Xu, Y; Mayne, L; Englander, SW. Evidence for an unfolding and refolding pathway in cytochrome c. Nat. Struct. Biol 1998, 5, 774–778. [Google Scholar]
- Bai, Y; Milne, JS; Mayne, L; Englander, SW. Protein stability parameters measured by hydrogen exchange. Proteins 1994, 20, 4–14. [Google Scholar]
- Chen, E; Van Vranken, V; Kliger, DS. The folding kinetics of the SDS-induced molten globule form of reduced cytochrome c. Biochemistry 2008, 47, 5450–5459. [Google Scholar]
- Takahashi, M; Yoshikawa, K; Vasilevskaya, VV; Khokhlov, AR. Discrete coil-globule transition of single duplex DNAs induced by polyamines. J. Phys. Chem. B 1997, 101, 9396–9401. [Google Scholar]
- Flory, PJ. Principles of Polymer Chemistry; Cornell University: Ithaca, NY, USA, 1953. [Google Scholar]
- Uversky, VN; Ptitsyn, OB. “Partly-folded” state — a new equilibrium state of protein molecules: Four-state guanidinium chloride-induced unfolding of β-lactamase at low temperature. Biochemistry 1994, 33, 2782–2791. [Google Scholar]
- Uversky, VN; Ptitsyn, OB. Further evidence on the equilibrium “pre-molten globule state”: Four-state guanidinium chloride-induced unfolding of carbonic anhydrase B at low temperature. J. Mol. Biol 1996, 255, 215–228. [Google Scholar]
- Matouscheck, A; Kellis, JT, Jr; Serrano, L; Fersht, AR. Mapping the transition state and pathway of folding by protein engineering. Nature 1989, 340, 122–126. [Google Scholar]
- Travaglini-Allocatelli, C; Gianni, S; Brunori, M. A common folding mechanism in the cytochrome c family. TIBS 2004, 29, 535–541. [Google Scholar]
- Travaglini-Allocatelli, C; Gianni, S; Morea, V; Tramontano, A; Soulimane, T; Brunori, M. Exploring the cytochrome c folding mechanism. J. Biol. Chem 2003, 278, 41136–41140. [Google Scholar]
- Travaglini-Allocatelli, C; Gianni, S; Dubey, VK; Borgia, A; Di Matteo, A; Bonivento, D; Cutruzzola, F; Bren, KL; Brunori, M. An obligatory intermediate in the folding pathway of cytochrome c552 from Hydrogenobacter thermophilus. J. Biol. Chem 2005, 280, 25729–25734. [Google Scholar]
- Borgia, A; Bonivento, D; Travaglini-Allocatelli, C; Di Matteo, A; Brunori, M. Unveiling a hidden folding intermediate in c-type cytochromes by protein engineering. J. Biol. Chem 2006, 281, 9331–9336. [Google Scholar]




© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goldbeck, R.A.; Chen, E.; Kliger, D.S. Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c. Int. J. Mol. Sci. 2009, 10, 1476-1499. https://doi.org/10.3390/ijms10041476
Goldbeck RA, Chen E, Kliger DS. Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c. International Journal of Molecular Sciences. 2009; 10(4):1476-1499. https://doi.org/10.3390/ijms10041476
Chicago/Turabian StyleGoldbeck, Robert A., Eefei Chen, and David S. Kliger. 2009. "Early Events, Kinetic Intermediates and the Mechanism of Protein Folding in Cytochrome c" International Journal of Molecular Sciences 10, no. 4: 1476-1499. https://doi.org/10.3390/ijms10041476