Construction of an Artificial MicroRNA Expression Vector for Simultaneous Inhibition of Multiple Genes in Mammalian Cells

Abstract
:1. Introduction
2. Results and Discussion
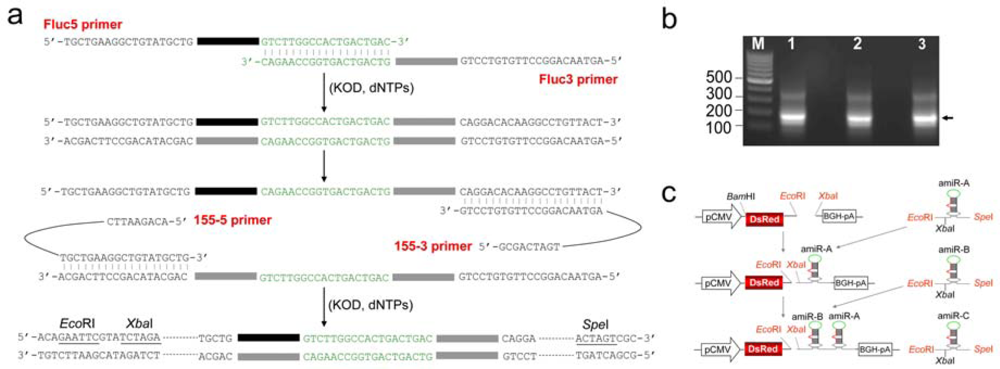
2.1. Construction of amiRNA expression vectors
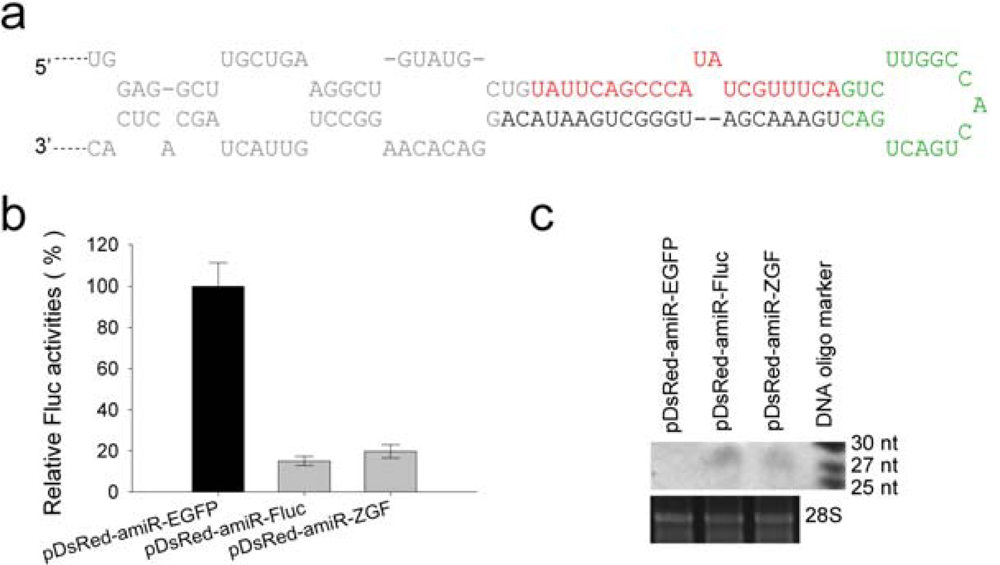
2.2. Suppression of firefly luciferase expression by amiR-Fluc expression vectors
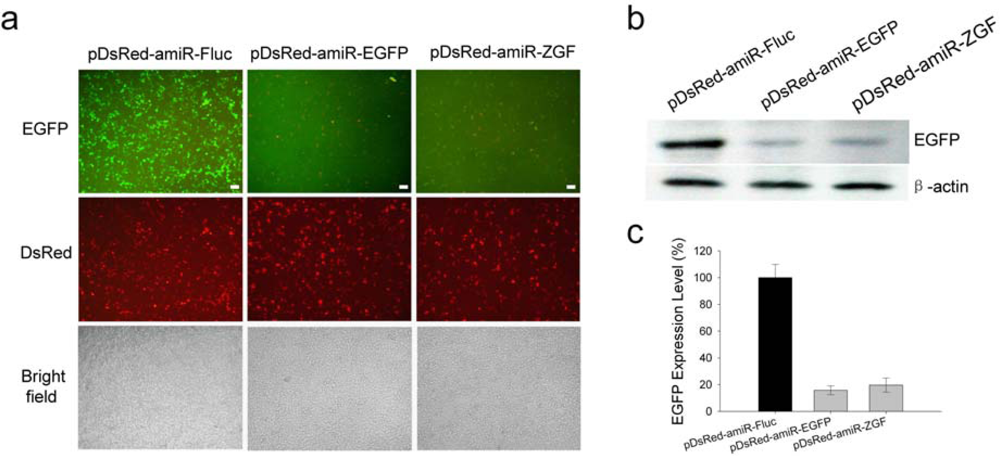
2.3. Efficient suppression of EGFP expression by amiR-EGFP expression vectors
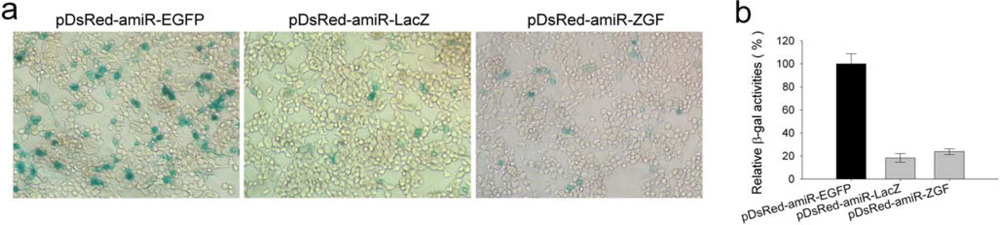
2.4. Efficient inhibition of β-galactosidase expression by amiR-LacZ expression vectors
3. Experimental Section
3.1. Synthesis of amiRNA cassette by one-step PCR
3.2. Plasmid construction
3.3. Cell culture and DNA transfection
3.4. Reporter assays
3.5. Western blotting
3.6. Northern blotting
4. Conclusions
Acknowledgments
References
- Bushati, N; Cohen, SM. MicroRNA functions. Annu. Rev. Cell Dev. Biol 2007, 23, 175–205. [Google Scholar]
- Lee, Y; Kim, M; Han, J; Yeom, KH; Lee, S; Baek, SH; Kim, VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004, 23, 4051–4060. [Google Scholar]
- Cai, X; Hagedorn, CH; Cullen, BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar]
- Kim, VN; Nam, JW. Genomics of microRNA. Trends Genet 2006, 22, 165–173. [Google Scholar]
- Suh, MR; Lee, Y; Kim, JY; Kim, SK; Moon, SH; Lee, JY; Cha, KY; Chung, HM; Yoon, HS; Moon, SY; Kim, VN; Kim, KS. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol 2004, 270, 488–498. [Google Scholar]
- Lee, Y; Ahn, C; Han, J; Choi, H; Kim, J; Yim, J; Lee, J; Provost, P; Radmark, O; Kim, S; Kim, VN. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar]
- Han, J; Lee, Y; Yeom, KH; Kim, YK; Jin, H; Kim, VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev 2004, 18, 3016–3027. [Google Scholar]
- Lund, E; Guttinger, S; Calado, A; Dahlberg, JE; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar]
- Yi, R; Qin, Y; Macara, IG; Cullen, BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003, 17, 3011–3016. [Google Scholar]
- Hutvagner, G; McLachlan, J; Pasquinelli, AE; Balint, E; Tuschl, T; Zamore, PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 2001, 293, 834–838. [Google Scholar]
- Ketting, RF; Fischer, SE; Bernstein, E; Sijen, T; Hannon, GJ; Plasterk, RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev 2001, 15, 2654–2659. [Google Scholar]
- Gregory, RI; Chendrimada, TP; Cooch, N; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar]
- Zeng, Y; Wagner, EJ; Cullen, BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol. Cell 2002, 9, 1327–1333. [Google Scholar]
- Sun, D; Melegari, M; Sridhar, S; Rogler, CE; Zhu, L. Multi-miRNA hairpin method that improves gene knockdown efficiency and provides linked multi-gene knockdown. Biotechniques 2006, 41, 59–63. [Google Scholar]
- Zhu, X; Santat, LA; Chang, MS; Liu, J; Zavzavadjian, JR; Wall, EA; Kivork, C; Simon, MI; Fraser, ID. A versatile approach to multiple gene RNA interference using microRNA-based short hairpin RNAs. BMC Mol. Biol 2007, 8, 98. [Google Scholar]
- Zhou, H; Huang, C; Xia, XG. A tightly regulated Pol III promoter for synthesis of miRNA genes in tandem. Biochim. Biophys. Acta 2008, 1779, 773–779. [Google Scholar]
- Du, G; Yonekubo, J; Zeng, Y; Osisami, M; Frohman, MA. Design of expression vectors for RNA interference based on miRNAs and RNA splicing. FEBS J 2006, 273, 5421–5427. [Google Scholar]
- Zhou, H; Xia, XG; Xu, Z. An RNA polymerase II construct synthesizes short-hairpin RNA with a quantitative indicator and mediates highly efficient RNAi. Nucleic Acids Res 2005, 33, e62. [Google Scholar]
- Zeng, Y; Cai, X; Cullen, BR. Use of RNA polymerase II to transcribe artificial microRNAs. Methods Enzymol 2005, 392, 371–380. [Google Scholar]
- Gou, D; Zhang, H; Baviskar, PS; Liu, L. Primer extension-based method for the generation of a siRNA/miRNA expression vector. Physiol. Genomics 2007, 31, 554–562. [Google Scholar]
- Chung, KH; Hart, CC; Al-Bassam, S; Avery, A; Taylor, J; Patel, PD; Vojtek, AB; Turner, DL. Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC/miR-155. Nucleic Acids Res 2006, 34, e53. [Google Scholar]
- Chang, K; Elledge, SJ; Hannon, GJ. Lessons from Nature: microRNA-based shRNA libraries. Nat. Methods 2006, 3, 707–714. [Google Scholar]
- Paddison, PJ; Cleary, M; Silva, JM; Chang, K; Sheth, N; Sachidanandam, R; Hannon, GJ. Cloning of short hairpin RNAs for gene knockdown in mammalian cells. Nat. Methods 2004, 1, 163–167. [Google Scholar]
- McIntyre, GJ; Fanning, GC. Design and cloning strategies for constructing shRNA expression vectors. BMC Biotechnol 2006, 6, 1. [Google Scholar]
- Saetrom, P; Snove, O; Nedland, M; Grunfeld, TB; Lin, Y; Bass, MB; Canon, JR. Conserved microRNA characteristics in mammals. Oligonucleotides 2006, 16, 115–144. [Google Scholar]
- Ritchie, W; Legendre, M; Gautheret, D. RNA stem-loops: to be or not to be cleaved by RNAse III. RNA 2007, 13, 457–462. [Google Scholar]
- Zeng, Y; Cullen, BR. Efficient processing of primary microRNA hairpins by Drosha requires flanking nonstructured RNA sequences. J. Biol. Chem 2005, 280, 27595–27603. [Google Scholar]
- Han, J; Lee, Y; Yeom, KH; Nam, JW; Heo, I; Rhee, JK; Sohn, SY; Cho, Y; Zhang, BT; Kim, VN. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar]
- Chen, CZ; Li, L; Lodish, HF; Bartel, DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar]
- Cullen, BR. Transcription and processing of human microRNA precursors. Mol. Cell 2004, 16, 861–865. [Google Scholar]
- Li, Z; Xiong, Y; Peng, Y; Pan, J; Chen, Y; Wu, X; Hussain, S; Tien, P; Guo, D. Specific inhibition of HIV-1 replication by short hairpin RNAs targeting human cyclin T1 without inducing apoptosis. FEBS Lett 2005, 579, 3100–3106. [Google Scholar]
- Hu, T; Fu, Q; Chen, P; Zhang, K; Guo, D. Generation of a stable mammalian cell line for simultaneous expression of multiple genes by using 2A peptide-based lentiviral vector. Biotechnol. Lett 2009, 31, 353–359. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5’-3’) |
|---|---|
| 155-5 | ACAGAATTCGACTCTAGAATCCTCTGGCTGCTGGAGGCTTGCTGAAGGCTGTATGCTG |
| 155-3 | GCGACTAGTACGGTGGCCATTTGTTCCATGTGAGTGCTAGTAACAGGCCTTGTGTCCTG |
| Fluc5 | TGCTGAAGGCTGTATGCTGTATTCAGCCCATATCGTTTCAGTCTTGGCCACTGACTGAC |
| Fluc3 | AGTAACAGGCCTTGTGTCCTGTATTCAGCCCATCGTTTCAGTCAGTCAGTGGCCAAGAC |
| EGFP5 | TGCTGAAGGCTGTATGCTGATGAACTTCAGGGTCAGCTTGGTCTTGGCCACTGACTGAC |
| EGFP3 | AGTAACAGGCCTTGTGTCCTGATGAACTTCAGTCAGCTTGGTCAGTCAGTGGCCAAGAC |
| LacZ5 | TGCTGAAGGCTGTATGCTGAAATCGCTGATTTGTGTAGTCGTCTTGGCCACTGACTGAC |
| LacZ3 | AGTAACAGGCCTTGTGTCCTGAAATCGCTGATGTGTAGTCGTCAGTCAGTGGCCAAGAC |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hu, T.; Fu, Q.; Chen, P.; Ma, L.; Sin, O.; Guo, D. Construction of an Artificial MicroRNA Expression Vector for Simultaneous Inhibition of Multiple Genes in Mammalian Cells. Int. J. Mol. Sci. 2009, 10, 2158-2168. https://doi.org/10.3390/ijms10052158
Hu T, Fu Q, Chen P, Ma L, Sin O, Guo D. Construction of an Artificial MicroRNA Expression Vector for Simultaneous Inhibition of Multiple Genes in Mammalian Cells. International Journal of Molecular Sciences. 2009; 10(5):2158-2168. https://doi.org/10.3390/ijms10052158
Chicago/Turabian StyleHu, Tao, Qiong Fu, Ping Chen, Li Ma, Onsam Sin, and Deyin Guo. 2009. "Construction of an Artificial MicroRNA Expression Vector for Simultaneous Inhibition of Multiple Genes in Mammalian Cells" International Journal of Molecular Sciences 10, no. 5: 2158-2168. https://doi.org/10.3390/ijms10052158