Trends in the Molecular Pathogenesis and Clinical Therapeutics of Common Neurodegenerative Disorders

 ,
,
Abstract
:1. Introduction
2. General Underlying Mechanisms Resulting in Neurodegeneration
2.1. Network dysfunction
2.2. Synaptic dysfunction leading to network failure
2.3. Survival of neurons
3. Overview of Alzheimer’s Disease
3.1. The neuropathology of Alzheimer’s disease
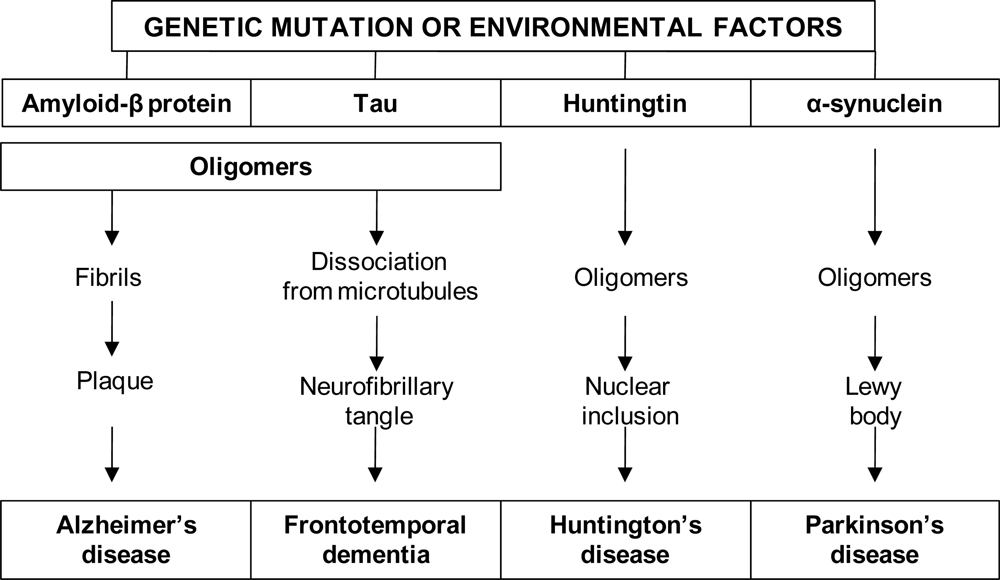
3.1.1. The amyloid hypothesis
3.1.2. The tau hypothesis
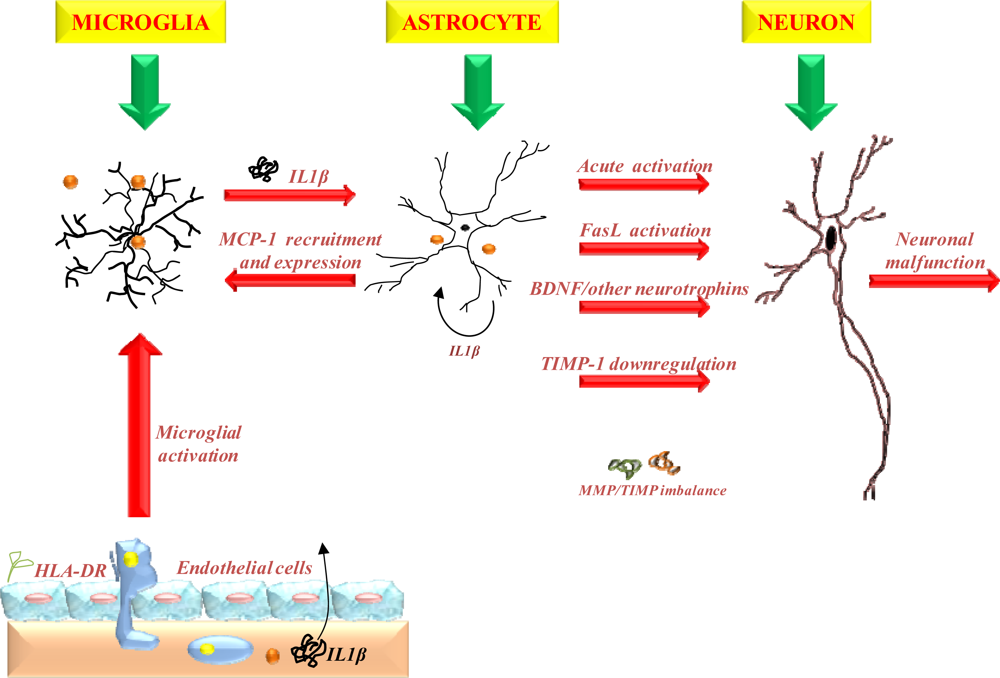
3.1.3. The role of inflammation
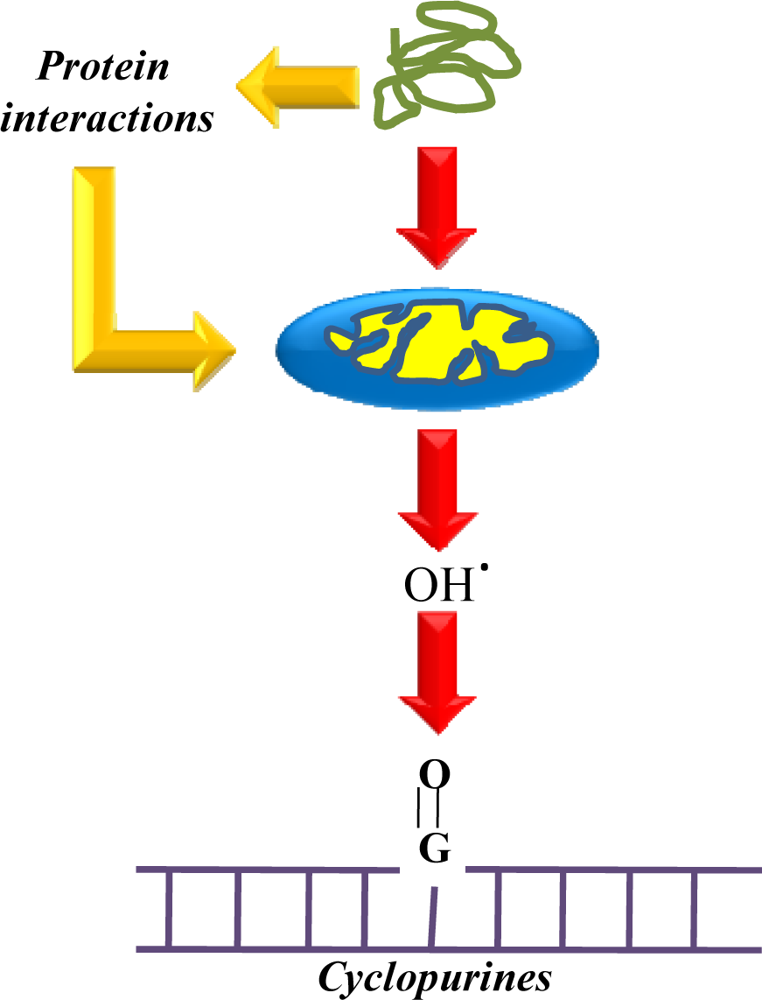
3.1.4. The role of free radicals
3.1.5. Excitotoxicity
3.1.6. Other risk factors
3.2. Clinical and therapeutic findings on Alzheimer’s disease
4. Overview of Parkinson’s Disease
4.1. The neuropathology of Parkinson’s disease
4.2. Clinical and therapeutic findings on Parkinson’s disease
4.3. The symptomatic treatment of Parkinson’s disease
5. Overview of Amyotrophic Lateral Sclerosis
5.1. The neuropathology of Amyotrophic Lateral Sclerosis: The role of inflammation
5.2. Clinical and therapeutic findings on Amyotrophic Lateral Sclerosis
5.3. The symptomatic treatment of Amyotrophic Lateral Sclerosis
6. Overview of Huntington’s Disease
6.1. The neuropathology of Huntington’s disease
6.2. The symptomatic treatment of Huntington’s disease
7. Other Related Atypical Neurodegenerative Disorders
8. The Therapeutic Implications of Neurodegenerative Disorders
9. Specialized Drug Delivery Strategies for the Treatment of Neurodegenerative Disorders
10. Conclusions
References
- Cowan, WM; Kandel, ER. Prospects for neurology and psychiatry. J. Am. Med. Assoc 2001, 285, 594–600. [Google Scholar]
- Broom, WJ; Parton, MJ; Vance, CA; Russ, C; Andersen, PM; Hansen, V; Leigh, PN; Powell, JF; Al-Chalabi, A; Shaw, CE. No association of the SOD1 locus and disease susceptibility or phenotype in sporadic ALS. Neurology 2004, 63, 2419–2422. [Google Scholar]
- Lefaucheur, JP. Motor cortex dysfunction revealed by cortical excitability studies in Parkinson’s disease: Influence of antiparkinsonian treatment and cortical stimulation. Clin. Neurophysiol 2005, 116, 244–253. [Google Scholar]
- Ahmed, N; Ahmed, U; Thornalley, PJ; Hager, K; Fleischer, G; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem 2005, 92, 255–263. [Google Scholar]
- Ivanoiu, A; Adam, S; Van der Linden, M; Salmon, E; Juillerat, A; Mulligan, R; Seron, X. Memory evaluation with a new cued recall test in patients with mild cognitive impairment and Alzheimer’s disease. J. Neurol 2005, 252, 47–55. [Google Scholar]
- Veldink, JH; Kalmijn, S; van den Berg, LH. Physical activity and the association with sporadic ALS. Neurol 2005, 64, 241–245. [Google Scholar]
- Lee, VM; Goedert, M; Trojanowski, JQ. Neurodegenerative tauopathies. Annu. Rev. Neurosci 2001, 24, 1121–1159. [Google Scholar]
- Riemenschneider, M; Klopp, N; Xiang, W; Wagenpfeil, S; Vollmert, C; Müller, U; Förstl, H; Illig, T; Kretzschmar, H; Kurz, A. Prion protein codon 129 polymorphism and risk of Alzheimer disease. Neurology 2004, 63, 364–366. [Google Scholar]
- Sengoku, R; Saito, Y; Ikemura, M; Hatsuta, H; Sakiyama, Y; Kanemaru, K; Arai, T; Sawabe, M; Tanaka, N; Mochizuki, H; Inoue, K; Murayama, S. Incidence and extent of Lewy body-related alpha-synucleinopathy in aging human olfactory bulb. J. Neuropathol. Exp. Neurol 2008, 67, 742–749. [Google Scholar]
- Matsuo, ES; Shin, RW; Billingsley, ML; Van deVoorde, A; O’Connor, M; Trojanowski, JQ; Lee, VM. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar]
- Lee, EM; Kim, JY; Cho, BR; Chung, WK; Yoon, BW; Kim, SU; Lee, BC; Hwang, WS; Moon, SY; Lee, JS; Ahn, C. Down-regulation of MHC class I expression in human neuronal stem cells using viral stealth mechanism. Biochem. Biophys. Res. Commun 2005, 326, 825–835. [Google Scholar]
- Shahani, N; Brandt, R. Functions and malfunctions of the tau proteins. Cell Mol Life Sci 2002, 59, 1668–1680. [Google Scholar]
- Rissman, RA; Rissman, RA; Poon, WW; Blurton-Jones, M; Oddo, S; Torp, R; Vitek, MP; LaFerla, FM; Rohn, TT; Cotman, CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Invest 2004, 14, 121–130. [Google Scholar]
- King, ME. Can tau filaments be both physiologically beneficial and toxic? Biochim. Biophys. Acta 2005, 1739, 260–267. [Google Scholar]
- Mattson, MP; Sherman, M. Perturbed signal transduction in neurodegenerative disorders involving aberrant protein aggregation. Neuro. Mol. Med 2003, 4, 109–131. [Google Scholar]
- Yuan, J; Yankner, BA. Apoptosis in the nervous system. Nature 2000, 407, 802–809. [Google Scholar]
- Morrison, JH; Hof, PR. Life and death of neurons in the aging brain. Sci 1997, 278, 412–419. [Google Scholar]
- Kordower, JH; Chu, Y; Stebbins, GT; DeKosky, ST; Cochran, EJ; Bennett, D; Mufson, EJ. Loss and atrophy of layer II entorhinal cortex neurons in elderly people with mild cognitive impairment. Ann. Neurol 2001, 49, 202–213. [Google Scholar]
- Blanchet, PJ. Antipsychotic drug-induced movement disorders. Can. J. Neurol. Sci 2003, 30, 101–107. [Google Scholar]
- Arendt, T. Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: the ‘Dr. Jekyll and Mr. Hyde concept’ of Alzheimer’s disease or the yin and yang of neuroplasticity. Prog. Neurobiol 2001, 71, 283–248. [Google Scholar]
- Palop, JJ; Chin, J; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar]
- Price, JL; Ko, AI; Wade, MJ; Tsou, SK; McKeel, DW; Morris, JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch. Neurol 2001, 58, 1395–1402. [Google Scholar]
- Greffard, S; Verny, M; Bonnet, AM; Beinis, JY; Gallinari, C; Meaume, S; Piette, F; Hauw, JJ; Duyckaerts, C. Motor score of the Unified Parkinson Disease Rating Scale as a good predictor of Lewy body-associated neuronal loss in the substantia nigra. Arch. Neurol 2006, 63, 584–588. [Google Scholar]
- Terry, RD; Masliah, E; Salmon, DP; Butters, N; DeTeresa, R; Hill, R; Hansen, LA; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol 1991, 30, 572–580. [Google Scholar]
- Honig, LS; Rosenberg, RN. Apoptosis and neurologic disease. Am. J. Med 2000, 108, 317–330. [Google Scholar]
- Onteniente, B. Natural and synthetic inhibitors of caspases: Targets for novel drugs. Curr. Drug Targ. CNS Neurol. Disord 2000, 3, 333–340. [Google Scholar]
- Gao, HM; Jiang, J; Wilson, B; Zhang, W; Hong, JS; Liu, B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J. Neurochem 2002, 81, 1285–1297. [Google Scholar]
- Calabrese, V; Scapagnini, G; Colombrita, C; Ravagna, A; Pennisi, G; Giuffrida, SAM; Galli, F; Butterfield, DA. Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: a nutritional approach. Amino Acids 2003, 25, 437–444. [Google Scholar]
- Miledi, R; Duen, Z; Martinez-Torres, A; Kawas, CH; Eusebi, F. Microtransplantation of functional receptors and channels from the Alzheimer’s brain to frog oocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 1760–1763. [Google Scholar]
- Hwang, DY; Cho, JS; Lee, SH; Chae, KR; Lim, HJ; Min, SH; Seo, SJ; Song, YS; Song, CW; Paik, SG; Sheen, YY; Kim, YK. Aberrant expressions of pathogenic phenotype in Alzheimer’s disease transgenic mice carrying NSE-controlled APPsw. Exp. Neurol 2004, 186, 20–32. [Google Scholar]
- Mani, RB. The evaluation of disease modifying therapies in Alzheimer’s disease: A regulatory viewpoint. Stat. Med 2004, 23, 305–314. [Google Scholar]
- Bosch, M; Pineda, JR; Suñol, C; Petriz, J; Cattaneo, E; Alberch, J; Canals, JM. Induction of GABAergic phenotype in a neural stem cell line for transplantation in an excitotoxic model of Huntington’s disease. Exp. Neurol 2004, 190, 42–58. [Google Scholar]
- Goldman, SA. Directed mobilization of endogenous neural progenitor cells: the intersection of stem cell biology and gene therapy. Curr. Opin. Mol. Ther 2004, 6, 466–472. [Google Scholar]
- Mohapel, P; Brundin, P. Harnessing endogenous stem cells to treat neurodegenerative disorders of the basal ganglia. Parkinsonism Relat. Disord 2004, 10, 259–264. [Google Scholar]
- Lewin, R. Is your brain really necessary? Science 1980, 210, 1232–1234. [Google Scholar]
- Chen, R; Cohen, LG; Hallett, M. Nervous system reorganization following injury. Neurosci 2002, 111, 761–773. [Google Scholar]
- Muchowski, PJ; Wacker, JL. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci 2005, 6, 11–22. [Google Scholar]
- Hynd, MR; Scott, HL; Dodd, PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int 2004, 45, 583–595. [Google Scholar]
- Wyss-Coray, T; Mucke, L. Inflammation in neurodegenerative disease: A double-edged sword. Neuron 2002, 35, 419–432. [Google Scholar]
- Beal, MF. Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 2005, 58, 495–505. [Google Scholar]
- Handley, OJ; Naji, JJ; Dunnett, SB; Rosser, AE. Pharmaceutical, cellular and genetic therapies for Huntington’s disease. Clin. Sci 2006, 110, 73–88. [Google Scholar]
- Levine, MS; Cepeda, C; Hickey, MA; Fleming, SM; Chesselet, MF. Genetic mouse models of Huntington’s and Parkinson’s diseases: illuminating but imperfect. Trends Neurosci 2004, 27, 691–697. [Google Scholar]
- Lazarov, O; Robinson, J; Tang, YP; Hairston, IS; Korade-Mirnics, Z; Lee, VM; Hersh, LB; Sapolsky, RM; Mirnics, K; Sisodia, SS. Environmental enrichment reduces A levels and amyloid deposition in transgenic mice. Cell 2005, 120, 701–713. [Google Scholar]
- Van Dellen, A; Grote, HE; Hannan, AJ. Gene–environment interactions, neuronal dysfunction and pathological plasticity in Huntington’s disease. Clin. Exp. Pharmacol. Physiol 2005, 32, 1007–1019. [Google Scholar]
- Mahley, RW; Weisgraber, KH; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl Acad. Sci 2006, 103, 5644–5651. [Google Scholar]
- Bezard, E; Gross, CE; Brotchie, JM. Presymptomatic compensation in Parkinson’s disease is not dopamine-mediated. Trends Neurosci 2003, 26, 215–221. [Google Scholar]
- Stern, Y. What is cognitive reserve? Theory and research application of the reserve concept. J. Int. Neuropsychol. Soc 2002, 8, 448–460. [Google Scholar]
- Buckner, RL. Memory and executive function in aging and AD: Multiple factors that cause decline and reserve factors that compensate. Neuron 2004, 44, 195–208. [Google Scholar]
- Maguire, EA; Valentine, ER; Wilding, JM; Kapur, N. Routes to remembering: the brains behind superior memory. Nat. Neurosci 2003, 6, 90–95. [Google Scholar]
- Zlokovic, BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 2005, 28, 202–208. [Google Scholar]
- Miller, DL; Ortega, S; Bashayan, O; Basch, R; Basilica, C. Compensation by fibroblast growth factor 1 (fgf1) does not account for the mild phenotypic defects observed in fgf2 null mice. Mol. Cell. Biol 2000, 20, 2260–2268. [Google Scholar]
- Dono, R; Texido, G; Dussel, R; Ehmke, H; Zeller, R. Impaired cerebral cortex development and blood pressure regulation in FGF-2-deficient mice. Embryo J 1998, 17, 4213–4225. [Google Scholar]
- Feldman, B; Poueymirou, W; Papaioannou, VE; DeChiara, TM; Goldfarb, M. Requirement of FGF-4 for post-implantation mouse development. Science 1995, 267, 246–249. [Google Scholar]
- Floss, T; Arnold, HH; Braun, T. A role for FGF-6 in skeletal muscle regeneration. Genes Dev 1997, 11, 2040–2051. [Google Scholar]
- Ortega, S; Ittmann, M; Tsang, SH; Ehrlich, M; Basilico, C. Neuronal defects and delayed wound healing in mice lacking fibroblast growth factor 2. Proc. Natl. Acad. Sci. USA 1998, 95, 5672–5677. [Google Scholar]
- Prinz, AA; Bucher, D; Marder, E. Similar network activity from disparate circuit parameters. Nat. Neurosci 2004, 7, 1345–1352. [Google Scholar]
- Whone, AL; Watts, RL; Stoessl, AJ; Davis, M; Reske, S; Nahmias, C; Lang, AE; Rascol, O; Ribeiro, MJ; Remy, P. Slower progression of Parkinsons disease with ropinirole versus levodopa. Ann. Neurol 2003, 54, 93–101. [Google Scholar]
- Evans, DA. Etimated prevalence of Alzheimer’s disease in the United States. Milbank Q 1990, 68, 267–289. [Google Scholar]
- Palop, JJ; Chin, J; Mucke, L. A network dysfunction perspective on neurodegenerative diseases. Nature 2006, 443, 768–773. [Google Scholar]
- Heckmann, JM; Low, WC; de Villiers, C; Rutherford, S; Vorster, A; Rao, H; Morris, CM; Ramesar, RS; Kalaria, RN. Novel presenilin 1 mutation with profound neurofibrillary pathology in an indigenous Southern African family with early-onset Alzheimer’s disease. Brain 2004, 127, 133–142. [Google Scholar]
- Vazey, K; Chen, S; Hughes, M; Connor, B. Transplanted adult neural progenitor cells survive, differentiate and reduce motor function impairment in a rodent model of Huntington’s disease. Exp. Neurol 2006, 199, 384–396. [Google Scholar]
- Ende, N; Chen, R. Human umbilical cord blood cells ameliorate Huntington’s disease in transgenic mice. J. Med 2001, 32, 231–240. [Google Scholar]
- Lescaudron, L; Unni, D; Dunbar, GL. Autologous adult bone marrow stem cell transplantation in an animal model of huntington’s disease: behavioral and morphological outcomes. Int. J. Neurosci 2003, 113, 945–956. [Google Scholar]
- Newman, MB; Davis, CD; Borlongan, CV; Emerich, D; Sanberg, PR. Transplantation of human umbilical cord blood cells in the repair of CNS diseases. Expert Opin. Biol. Ther 2004, 4, 121–130. [Google Scholar]
- Ferri, CP; Prince, M; Brayne, C; Brodaty, H; Fratiglioni, L; Ganguli, M; Hall, K; Hasegawa, K; Hendrie, H; Huang, Y; Jorm, A; Mathers, C; Menezes, PR; Rimmer, E; Scazufca, M. Global prevalence of dementia: A Delphi consensus study. Lancet 2005, 366, 2112–2117. [Google Scholar]
- Andlin-Sobocki, P; Jönsson, B; Wittchen, HU; Olesen, J. Cost of disorders of the brain in Europe. Eur. J. Neurol 2005, 12, 1–27. [Google Scholar]
- Jönsson, L; Berr, C. Cost of dementia in Europe. Eur. J. Neurol 2005, 12, 50–53. [Google Scholar]
- Terry, RD; Katzman, R. Senile dementia of the Alzheimer type. Ann. Neurol 1983, 14, 497–506. [Google Scholar]
- Kennedy, JL; Farrer, LA; Andreasen, NC; Mayeux, R; St George-Hyslop, P. The genetics of adult-onset neuropsychiatric disease: Complexities and conundra? Science 2003, 302, 822–826. [Google Scholar]
- McKhann, G; Drachman, D; Folstein, M; Katzman, R; Price, D; Stadlan, EM. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurol 1984, 34, 939–944. [Google Scholar]
- Galasko, D; Hansen, LA; Katzman, R; Wiederholt, W; Masliah, E; Terry, R; Hill, LR; Lessin, P; Thal, LJ. Clinical neuropathological correlations in Alzheimer’s disease and related dementias. Arch. Neurol 1994, 51, 888–895. [Google Scholar]
- Kosunen, O; Soininen, H; Paljärvi, L; Heinonen, O; Talasniemi, S; Riekkinen, PJ. Diagnostic accuracy of Alzheimer’s disease: a neuropathological study. Acta Neuropathol (Berl) 1991, 185–193. [Google Scholar]
- Blennow, K; Hampel, H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol 2003, 2, 605–613. [Google Scholar]
- Cummings, JL; Vinters, HV; Cole, GM; Khachaturian, ZS. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51, 2–17. [Google Scholar]
- Larson, EB; Edwards, JK; O’Meara, E; Nochlin, D; Sumi, SM. Neuropathologic diagnostic outcomes from a cohort of outpatients with suspected dementia. J. Gerontol. A Biol. Sci. Med. Sci 1996, 51, 313–318. [Google Scholar]
- Rasmusson, DX; Brandt, J; Steele, C; Hedreen, JC; Troncoso, JC; Folstein, MF. Accuracy of clinical diagnosis of Alzheimer disease and clinical features of patients with non-Alzheimer disease neuropathology. Alzheimer Dis. Assoc. Disord 1996, 10, 180–188. [Google Scholar]
- American Psychiatric Association DSM-IV. Diagnostic and Statistical Manual, 4th Ed ed; American Psychiatric Association: Washington, DC, USA, 1994. [Google Scholar]
- International Classification of Diseases (ICD-10). Classification of Mental and Behavioural Disorders, 10th Ed ed; World Health Organization: Geneva, Switzerland, 1992. [Google Scholar]
- Petersen, RC. Mild cognitive impairment: transition between aging and Alzheimer’s disease. Neurologia 2000, 15, 93–101. [Google Scholar]
- Petersen, RC; Smith, GE; Ivnik, RJ; Tangalos, EG; Schaid, DJ; Thibodeau, SN; Kokmen, E; Waring, SC; Kurland, LT. Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. JAMA 1995, 273, 1274–1278. [Google Scholar]
- Petersen, RC; Smith, GE; Waring, SC; Ivnik, RJ; Tangalos, EG; Kokmen, E. Mild cognitive impairment: clinical characterization and outcome. Arch. Neurol 1999, 56, 303–308. [Google Scholar]
- Rocchi, A; Pellegrini, S; Siciliano, G; Murri, L. Causative and susceptibility genes for Alzheimer’s disease: A review. Brain Res. Bull 2003, 61, 1–24. [Google Scholar]
- Niewöhner, J; Tannert, C. Gene therapy: Prospective technology assessment in its societal context, 1st ed; Elsevier Science: Berlin, Germany, 2006. [Google Scholar]
- Peterson, J. Immune deficiency and balance disorder result from a single gene defect. Available online: http://www.jax.org/news/balance_gene_defect.html, accessed 2008.
- Martins, IC; Kuperstein, I; Wilkinson, H; Maes, E; Vanbrabant, M; Jonckheere, W; Van Gelder, P; Hartmann, D; D’Hooge, R; De Strooper, B; Schymkowitz, J; Rousseau, F. Lipids revert inert Aβ-amyloid fibrils to neurotoxic protofibrils that affect learning in mice. EMBO J 2008, 27, 224–233. [Google Scholar]
- Cummings, JL; Vinters, HV; Cole, GM; Khachaturian, ZS. Alzheimer’s disease: etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology 1998, 51, S2–S17. [Google Scholar]
- Schwartz, AL; Ciechanover, A. The ubiquitin–proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med 1999, 50, 57–74. [Google Scholar]
- Gotz, J; Tolnay, M; Barmettlet, R; Chen, F; Probst, A; Nitsch, RM. Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur. J. Neurosci 2001, 13, 2131–2140. [Google Scholar]
- Lamb, BT; Bardel, KA; Kulnane, LS; Anderson, JJ; Holtz, G; Wagner, SL; Sisodia, SS; Hoeger, EJ. Amyloid production and deposition in mutant amyloid presursor protein and presenilin-1 yeast artificial chromosome transgenic mice. Nat. Neurosci 1999, 2, 695–697. [Google Scholar]
- Selkoe, DJ; Schenk, D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Ann. Rev. Pharmacol. Toxicol 2003, 43, 545–584. [Google Scholar]
- Lleo, A; Blesa, R; Queralt, R; Ezquerra, M; Molinuevo, JL; Pena-Casanova, J; Rojo, A; Oliva, R. Frequency of mutations in the presenilin and amyloid precursor protein genes in early-onset Alzheimer disease in Spain. Arch. Neurol 2002, 59, 1759–1763. [Google Scholar]
- Cataldo, AM; Peterhoff, CM; Schmidt, SD; Terio, NB; Duff, K; Beard, M; Mathews, PM; Nixon, RA. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J. Neuropathol. Exp. Neurol 2004, 63, 821–830. [Google Scholar]
- Grundke-Iqbal, I; Iqbal, K. Tau pathology generated by over expression of tau. Am. J. Pathol 1999, 155, 1781–1785. [Google Scholar]
- Kosik, KS; Crandall, JE; Mufson, EJ; Neve, RL. Tau in situ hybridization in normal and Alzheimer brain: localization in the somatodendritic compartment. Ann. Neurol 1989, 26, 352–361. [Google Scholar]
- Papasozomenos, SC. Tau protein immunoreactivity in dementia of the Alzheimer type. I. Morphology, evolution, distribution, and pathogenetic implications. Lab Invest 1989, 60, 123–137. [Google Scholar]
- Brandt, R; Hundelt, M; Shahani, N. Tau alteration and neuronal degeneration in tauopathies: mechanisms and models. Biochim. Biophys. Acta 2005, 1739, 331–354. [Google Scholar]
- LaFerla, FM; Oddo, S. Alzheimer’s disease: Abeta, tau and synaptic dysfunction. Trends Mol. Med 2005, 11, 170–176. [Google Scholar]
- Braak, H; Braak, E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol 1991a, 1, 213–216. [Google Scholar]
- Braak, H; Braak, E. Neuropathological staging of Alzheimer-related changes. Acta. Neuropathol. (Berl) 1991, 82, 239–259. [Google Scholar]
- Shen, ZX. Brain cholinesterases: I. The clinico-histopathological and biochemical basis of Alzheimer’s disease. Med. Hypoth 2004, 63, 285–297. [Google Scholar]
- Selkoe, DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar]
- Zhang, Y; Hayes, A; Pritchard, A; Thaker, U; Haque, MS; Lemmon, H; Harris, J; Cumming, A; Lambert, J; Chartier-Harlin, M; St. Clair, D; Iwatsubo, T; Mann, DM; Lendon, CL. Interleukin-6 promoter polymorphism: risk and pathology of Alzheimer’s disease. Neurosci. Lett 2004, 362, 99–101. [Google Scholar]
- Luchsinger, JA; Mayeux, R. Dietary factors and Alzheimer’s disease. Lancet Neurol 2004, 3, 579–587. [Google Scholar]
- Schutte, DL. The evolving role of genomics in shaping care for persons with dementia. Nurs. Clin. North Am 2004, 39, 581–592. [Google Scholar]
- Souder, E; Beck, C. Overview of Alzheimer’s disease. Nurs. Clin. North Am 2004, 39, 545–559. [Google Scholar]
- Annaert, W; De Strooper, B. A cell biological perspective on Alzheimer’s disease. Ann. Rev. Cell Dev. Biol 2002, 18, 25–51. [Google Scholar]
- Tuppo, EE; Arias, HR. The role of inflammation in Alzheimer’s disease. Int. J. Biochem. Cell Biol 2005, 37, 289–305. [Google Scholar]
- Akiyama, H; Barger, S; Barnum, S; Bradt, B; Bauer, J; Cole, GM; Cooper, NR; Eikelenboom, P; Emmerling, M; Fiebich, BL; Finch, CE; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar]
- Liu, B; Hong, JS. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther 2003, 304, 1–7. [Google Scholar]
- Landi, F; Matteo, C; Graziano, O; Andrea, R; Sergio, T; Roberto, B. Non-Steroidal Anti-Inflammatory Drug (NSAID) use and Alzheimer disease in community-dwelling elderly patients. Am. J. Geriatr. Psych 2003, 11, 179–185. [Google Scholar]
- Bale, KR. Neurodegenerative disease research in the 21st century. Drug Disc. Today 2004, 9, 553–556. [Google Scholar]
- Rogers, JT; Lahiri, DK. Metal and inflammatory targets for Alzheimer’s disease. Curr. Drug Targ 2004, 5, 535–551. [Google Scholar]
- Ghorpade, A. To investigate the role of glial cells in central nervous system inflammation and neurodegeneration. University of Nebraska Medical Center, Department of Pharmacology and Experimental Neuroscience ( http://www.unmc.edu/dept/pharmacology/index.cfm?CONREF=68).
- Dhar, A; Gardner, J; Borgmann, K; Wu, L; Ghorpade, A. Novel role of TGF-β in differential astrocyte-TIMP-1 regulation: Implications for HIV-1-dementia and neuroinflammation. J. Neurosci. Res 2006, 83, 1271–1280. [Google Scholar]
- Dalle-Donne, I; Giustarini, D; Colombo, R; Rossi, R; Milzani, A. Protein carbonylation in human diseases. Trends Mol. Med 2003, 9, 169–176. [Google Scholar]
- Mariani, E; Polidori, MC; Cherubini, A; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 2005, 827, 65–75. [Google Scholar]
- Praticò, D. Peripheral biomarkers of oxidative damage in Alzheimer’s disease: The road ahead. Neurobiol. Aging 2005, 26, 581–583. [Google Scholar]
- Sayre, LM; Moreira, PI; Smith, MA; Perry, G. Metal ions and oxidative protein modification in neurological disease. Ann. Ist. Super. Sanita 2005, 41, 143–164. [Google Scholar]
- Hynd, MR; Scott, HL; Dodd, PR. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int 2004, 45, 583–595. [Google Scholar]
- Siesjö, BK. Cell damage in the brain: A speculative synthesis. J. Cereb. Blood Flow Metab 1981, 1, 155–185. [Google Scholar]
- Paschen, W. Role of calcium in neuronal cell injury: Which subcellular compartment is involved? Brain Res. Bull 2000, 53, 409–413. [Google Scholar]
- Katayama, T; Imaizumi, K; Manabe, T; Hitomi, J; Kudo, T; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat 2004, 28, 67–78. [Google Scholar]
- Schröder, M; Kaufman, RJ. ER stress and the unfolded protein response. Mutat. Res 2005, 569, 29–63. [Google Scholar]
- Rego, AC; Oliveira, CR. Mitochondrial dysfunction and reactive oxygen species in excitotoxicity and apoptosis: implications for the pathogenesis of neurodegenerative diseases. Neurochem. Res 2003, 28, 1563–1574. [Google Scholar]
- LaFerla, FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci 2002, 3, 862–872. [Google Scholar]
- Pereira, C; Agostinho, P; Moreira, PI; Cardoso, SM; Oliveira, CR. Alzheimer’s disease associated neurotoxic mechanisms and neuroprotective strategies. Curr. Drug Targ. CNS Neurol. Disord 2005, 4, 383–403. [Google Scholar]
- Hamanaka, H; Katoh-Fukui, Y; Suzuki, K; Kobayashi, M; Suzuki, R; Motegi, Y; Nakahara, Y; Takeshita, A; Kawai, M; Ishiguro, K; Yokoyama, M; Fujita, SC. Altered cholesterol metabolism in human apolipoprotein E4 knock-in mice. Human Mol. Gen 2000, 9, 353–361. [Google Scholar]
- Combarros, O; Alvarez-Arcaya, A; Sánchez-Guerra, M; Infante, J; Berciano, J. Candidate gene association studies in sporadic Alzheimer’s disease. Dement Geriatr Cogn Disord 2002, 14, 41–54. [Google Scholar]
- Loo, DT; Copani, A; Pike, CJ; Whittemore, ER; Walencewicz, AJ; Cotman, CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar]
- Su, JH; Anderson, AJ; Cummings, BJ; Cotman, CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 1994, 5, 2529–2533. [Google Scholar]
- Smale, G; Nichols, NR; Brady, DR; Finch, CE; Horton, WE, Jr. Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol 1995, 133, 225–230. [Google Scholar]
- Sastry, PS; Rao, KS. Apoptosis and the nervous system. J. Neurochem 2000, 74, 1–20. [Google Scholar]
- Chang, WP; Chang, W; Koelsch, G; Wong, S; Downs, D; Huining, DA; Weerasena, V; Gordon, B; Devasamudram, T; Bilcer, G; Ghosh, AK; Tang, J. In vivo inhibition of Aβ production by Memapsin 2 (β-secretase) inhibitors. J. Neurochem 2004, 89, 1409–1416. [Google Scholar]
- Jorda, EG; Verdaguer, E; Jiménez, A; Canudas, AM; Rimbau, V; Camps, P; Muñoz-Torrero, D; Camins, A; Pallàs, M. (+/–)-huprine Y, (–)-huperzine A and tacrine do not show neuroprotective properties in an apoptotic model of neuronal cytoskeletal alteration. J. Alzheimers Dis 2004, 6, 577–583. [Google Scholar]
- Farlow, MR; Lilly, ML. Rivastigmine: An open-label, observational study of safety and effectiveness in treating patients with Alzheimer’s disease for up to 5 years. BMC Geriatr 2005, 5, 3. [Google Scholar]
- National Institute on Aging (NIA). Evaluation of galantamine in the treatment of AD. Available online: http://clinicaltrials.gov/ct2/show/NCT00000172?term=Alzheimer's+Disease&rank=51, accessed 2008.
- Masterman, D. Cholinesterase inhibitors in the treatment of Alzheimer’s disease and related dementias. Clin. Geriatr. Med 2004, 20, 59–68. [Google Scholar]
- Schneider, LS. AD2000: donepezil in Alzheimer’s disease. Lancet 2004, 363, 2100–2101. [Google Scholar]
- Reisberg, B; Doody, R; Stöffler, A; Schmitt, F; Ferris, S; Jörg-Möbius, H. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med 2003, 348, 1333–1341. [Google Scholar]
- Winblad, B; Poritis, N. Memantine in severe dementia: results of the 9M-BEST Study (benefit and efficacy in severely demented patients during treatment with memantine). 1999, 14, 135–146. [Google Scholar]
- Rosenberg, PB. Clinical aspects of inflammation in Alzheimer’s disease. Int. Rev. Psychiatry 2005, 17, 503–514. [Google Scholar]
- Tabira, T. Vaccination therapy for Alzheimer’s disease. Brain Nerve 2007, 59, 375–382. [Google Scholar]
- Schenk, D; Seubert, P; Ciccarelli, B. Immunization with amyloid- beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar]
- Jeyarasasingam, G; Tompkins, L; Quik, M. Stimulation of non-alpha7 nicotinic receptors partially protects dopaminergic neurons from 1-methyl-4-phenylpyridinium-induced toxicity in culture. Neuroscience 2002, 109, 275–285. [Google Scholar]
- Conley, SC; Kirchner, JT. Parkinson’s disease-the shaking palsy, underlying factors, diagnostic considerations, and clinical course. Postgrad. Med 1999, 106, 39–42. [Google Scholar]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci 2001, 2, 492–501. [Google Scholar]
- Hashimoto, M; Kawahara, K; Bar-On, P; Rockenstein, E; Crews, L; Masliah, E. The role of α-synuclein assembly and metabolism in the pathogenesis of lewy body disease. J. Mol. Neurosci 2004, 24, 343–352. [Google Scholar]
- Hofer, A; Gasser, T. New aspects of genetic contributions to Parkinson’s disease. J. Mol. Neurosci 2004, 24, 417–424. [Google Scholar]
- Schulz, JB; Lindenau, J; Seyfried, J; Dichgans, J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem 2001, 267, 4904–4911. [Google Scholar]
- Spillantini, MG; Schmidt, ML; Lee, VMY; Trojanowski, JQ; Jakes, R; Goedert, M. α-synuclein in lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Wirths, O; Bayer, TA. Alpha-synuclein, Abeta and Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psych 2003, 27, 103–108. [Google Scholar]
- Snyder, H; Wolozin, B. Pathological proteins in Parkinson’s disease: focus on the proteasome. J. Mol. Neurosci 2004, 24, 425–442. [Google Scholar]
- Nobutaka, N; Yoshikun, M. Familial Parkinson’s disease based on the single gene defects. A key to the understanding the pathogenesis for nigral degeneration in sporadic Parkinson’s disease. Juntendo Med. J 2001, 47, 53–70. [Google Scholar]
- Braak, H; Del-Tredici, K; Rub, U; de Vos, RA; Jansen-Steur, EN; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar]
- Jellinger, KA; Mizuno, Y. Parkinson disease. In Neurodegeneration and Dementias; Dickson, DW, Ed.; ISN Press: Los Angeles, USA, 2003. [Google Scholar]
- Bernheimer, H; Birkmayer, W; Hornykiewicz, O; Jellinger, K; Seitelberger, F. Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci 1973, 20, 415–455. [Google Scholar]
- Rinne, JO; Nurmi, E; Ruottinen, HM; Bergman, J; Eskola, O; Solin, O. F-18FDOPA and F-18CFT are both sensitive PET markers to detect presynaptic dopaminergic hypofunction in early Parkinson’s disease. Synapse 2001, 40, 193–200. [Google Scholar]
- Blandini, F; Nappi, G; Tassorelli, C; Martignoni, E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog. Neurobiol 2000, 62, 63–88. [Google Scholar]
- Chase, TN; Oh, JD. Striatal mechanisms and pathogenesis of parkinsonian signs and motor complications. Ann. Neurol 2000, 47, 122–129. [Google Scholar]
- Bezard, E; Brotchie, JM; Gross, CE. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci 2001, 2, 577–588. [Google Scholar]
- Parent, A; Parent, M; Levesque, M. Basal ganglia and Parkinson disease: An anatomical perspective. Neurosci. News 1999, 2, 19–26. [Google Scholar]
- Freeman, A; Ciliax, B; Bakay, R; Daley, J; Miller, RD; Keating, G; Levey, A; Rye, D. Nigrostriatal collaterals to thalamus degenerate in parkinsonian animal models. Ann. Neurol 2001, 50, 321–329. [Google Scholar]
- Deuschl, G; Volkmann, J. Tremors: Differential Diagnosis, Pathophysiology, and Therapy. In Parkinson’s Disease and Movement Disorders, 4th Ed; Jankovic, JJ, Tolosa, E, Eds.; Lippincott Williams and Wilkins: Philadelphia, USA, 2002; pp. 270–290. [Google Scholar]
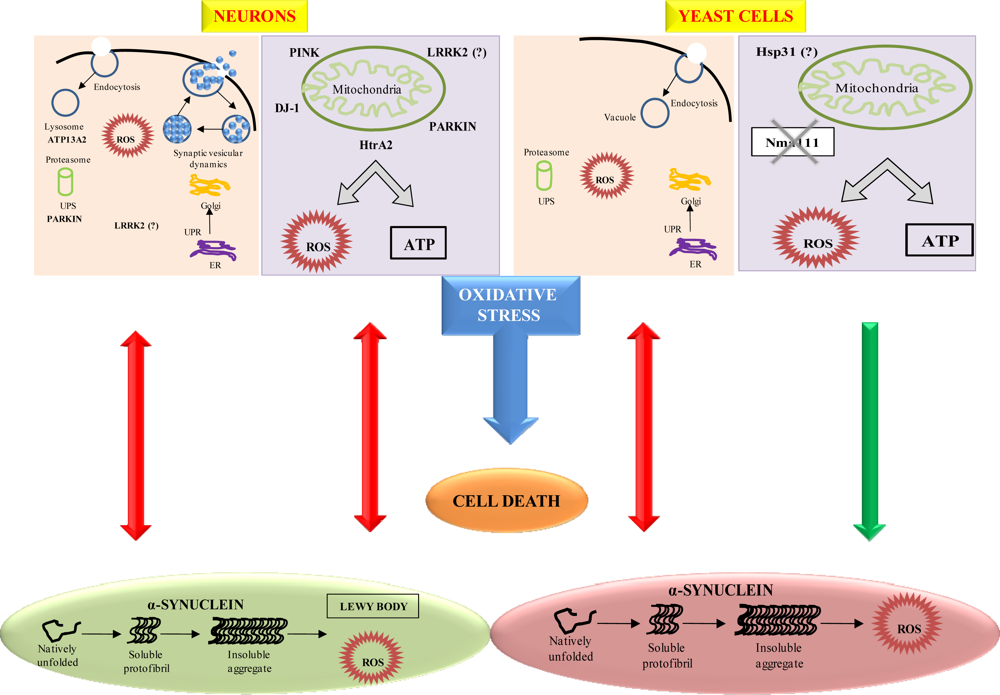
- Winderickx, J; Delay, C; De Vos, A; Klinger, H; Pellens, K; Vanhelmont, T; Van Leuven, F; Zabrocki, P. Protein folding diseases and neurodegeneration: Lessons learned from yeast. Mol. Cell Res 2008, 1783, 1381–1395. [Google Scholar]
- Bartzokis, G; Tishler, TA; Shin, IS; Lu, PH; Cummings, JL. Brain ferritin iron as a risk factor for age at onset in neurodegenerative diseases. Ann. New York Acad. Sci 2004, 1012, 224–236. [Google Scholar]
- Bridge, MH; Williams, E; Lyons, ME; Tipton, KF; Linert, W. Electrochemical investigation into the redox activity of Fe(II)/Fe(III) in the presence of nicotine and possible relations to neurodegenerative diseases. Biochim. Biophys. Acta 2004, 690, 77–84. [Google Scholar]
- Ostrerova-Golts, N; Petrucelli, L; Hardy, J; Lee, JM; Farer, M; Wolozin, B. The A53T α-synuclein mutation increases iron-dependent aggregation and toxicity. J. Neurosci 2000, 20, 6048–6054. [Google Scholar]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 2000, 18, 685–716. [Google Scholar]
- Outeiro, TF; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar]
- McLean, PJ; Kawamata, H; Hyman, BT. Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome-sensitive inclusions in primary neurons. Neuroscience 2001, 104, 901–912. [Google Scholar]
- Jo, E; McLaurin, J; Yip, CM; St. George-Hyslop, P; Fraser, PE. α-synuclein membrane interactions and lipid specificity. J. Biol. Chem 2000, 275, 34328. [Google Scholar]
- Seibyl, J; Jennings, D; Tabamo, R; Marek, K. Neuroimaging trials of Parkinson’s disease progression. J. Neurol 2004, 251, 9–13. [Google Scholar]
- Simuni, T; Hurtig, H. Levodopa: 30 years of progress. In Parkinson’s Disease Diagnosis and Clinical Management; Factor, SA, Weiner, WJ, Eds.; Demos Medical Publishing, Inc: Philadelphia, USA, 2000. [Google Scholar]
- Albin, RL; Frey, KA. Initial agonist treatment of Parkinson disease. Neurol 2003, 60, 390–394. [Google Scholar]
- Mercuri, NB; Benardi, G. The ‘magic’ of L-dopa: Why is it the gold standard Parkinson’s disease therapy? Trends Pharmacol. Sci 2005, 26, 341–344. [Google Scholar]
- Thanvi, BR; Lo, TC. Long term motor complications of levodopa: Clinical features, mechanisms, and management strategies. Postgrad. Med. J 2004, 80, 452–458. [Google Scholar]
- Woitalla, D; Müller, T; Benz, S; Horowski, R; Przuntek, H. Transdermal lisuride delivery in the treatment of Parkinson’s disease. J. Neural. Transm. Suppl 2004, 68, 89–95. [Google Scholar]
- Siddiqui, MA; Plosker, GL. Rasagiline. Drugs Aging 2005, 22, 83–91. [Google Scholar]
- Kim, MS; Lee, JI; Lee, WY; Kim, SE. Neuroprotective effect of Ginkgo biloba L. extract in a rat model of Parkinson’s disease. Phytother. Res 2004, 18, 663–666. [Google Scholar]
- Mochizuki, H. Future of gene therapy for Parkinson’s disease. Rinsho Shinkeigaku 2004, 44, 948–950. [Google Scholar]
- Levy, YS; Stroomza, M; Melamed, E; Offen, D. Embryonic and adult stem cells as a source for cell therapy in Parkinson’s disease. J. Mol. Neurosci 2004, 24, 353–386. [Google Scholar]
- Karimi and Eftekharpour. Fehlings lab, McEwen Centre for Regenerative Medicine. Available online: www.mcewencentre.com/res_prog_scnd.asp, accessed 2009.
- O’Brien, JT; Fenwick, J; Williams, ED; Firbank, M; Aarsland, D; McKeith, IG. Dopamine transporter loss visualized with FP-CIT SPECT in the differential diagnosis of dementia with lewy bodies. Arch. Neurol 2004, 61, 919–925. [Google Scholar]
- McKeith, I; O’Brien, J; Walker, Z; Tatsch, K. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol 2004, 6, 305–313. [Google Scholar]
- Walkinshaw, G; Waters, CM. Induction of apoptosis in catecholaminergic PC12 cells by L-DOPA. Implications for the treatment of Parkinson’s disease. J. Clin. Invest 1995, 95, 2458–2464. [Google Scholar]
- Whone, AL; Moore, RY; Piccini, PP; Brooks, DJ. Plasticity of the nigropallidal pathway in Parkinson’s disease. Ann. Neurol 2003, 53, 206–213. [Google Scholar]
- Brooks, DJ; Torjanski, N; Burn, DJ. Ropinirole in the symptomatic treatment of Parkinson’s disease. J. Neural. Transm. Suppl 1995, 45, 231–238. [Google Scholar]
- Uitti, RJ; Rajput, AH; Ahlskog, JE; Offord, KP; Schroeder, DR; Ho, MM; Prasad, M; Rajput, A; Basran, P. Amantadine treatment is an independent predictor of improved survival in Parkinson’s disease. Neurology 1996, 46, 1551–1556. [Google Scholar]
- Ebadi, MS; Pfeiffer, R. Amantadine neuroprotective effect on PD. Parkinson’s Dis 2005, 686. [Google Scholar]
- Münchau, A; Bhatia, KP. Pharmacological treatment of Parkinson’s disease. Postgrad. Med. J 2000, 76, 602–610. [Google Scholar]
- Oertel, WH; Quinn, NP. Neurological Disorders Course and Treatment. In Parkinsonism; Brandt, T, Caplan, LR, Dichgans, J, Eds.; Academic Press: San Diego, USA, 1996; pp. 715–772. [Google Scholar]
- Parkinson Study Group. Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Ann. Neurol 1997, 42, 747–755. [Google Scholar]
- Churchyard, A; Mathias, CJ; Boonkonchuen, P; Lees, A. Autonomic effects of selegeline: Possible cardiovascular toxicity in Parkinson’s disease. J Neurol Neurosurg Psychiatry 1997, 63, 228–234. [Google Scholar]
- Duvoisin, RC. Cholinergic-anticholinergic antagonisms in parkinsonism. Arch Neurol 1967, 17, 124–136. [Google Scholar]
- Pondal, M; Del Ser, T; Berjemo, F. Anticholinergic therapy and dementia in patients with Parkinson’s disease. J. Neurol 1996, 243, 543–546. [Google Scholar]
- Kaakkola, S; Gordin, A; Männisto, PT. General properties and clinical possibilities of new selective inhibitors of catechol-o-methyltransferase. Gen. Pharmacol 1994, 25, 813–824. [Google Scholar]
- Keräenen, T; Gordin, A; Harjola, VP; Karlsson, M; Korpela, K; Pentikäinen, PJ; Rita, H; Seppälä, L; Wikberg, T. The effect of catechol-O-methyltransferase inhibition by entacapone on the pharmacokinetics and metabolism of levodopa in healthy volunteers. Clin. Neuropharmacol 1993, 16, 145–156. [Google Scholar]
- Shults, CW; Oakes, D; Kieburtz, K; Beal, F; Haas, R; Plumb, S; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol 2002, 59, 1541–1550. [Google Scholar]
- Freed, CR; Greene, PE; Breeze, RE; Curt, R; Tsai, W; DuMouchel, W; Kao, R; Dillon, S; Winfield, H; Culver, S; Trojanowski, JQ; Eidelberg, D; Fahn, S. Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N. Engl. J. Med 2001, 344, 710–719. [Google Scholar]
- Kopyov, OV; Jacques, DS; Lieberman, A; Duma, CM; Rogers, RL. Outcome following intrastriatal fetal mesencephalic grafts for Parkinson’s patients is directly related to the volume of grafted tissue. Exp. Neurol 1997, 146, 536–545. [Google Scholar]
- Lopez-Lozano, J; Bravo, G; Brera, B; Dargallo, J; Salmeán, J; Uría, J; Insausti, J; Millán, I. Long-term follow-up in 10 Parkinson’s disease patients subjected to fetal brain grafting into a cavity in the caudate nucleus: The Clinica Puerta de Hierro experience. CPH Neural Transplantation Group. Transplant Proc 1995, 27, 1395–1400. [Google Scholar]
- Olanow, CW; Goetz, CG; Kordower, JH; Stoessl, AJ; Sossi, V; Brin, MF; Shannon, KM; Nauert, GM; Perl, DP; Godbold, J; Freeman, TB. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol 2003, 54, 403–414. [Google Scholar]
- McGuire, D; Garrison, L; Miller, RG. Relationship of the Tufts Quantitative Neuromuscular Exam(TQNE) and the Sickness Impact Profile (SIP) in measuring progression of ALS. Neurol 1996, 46, 1442–1444. [Google Scholar]
- Ritchie, K; Kildea, D. Is senile dementia “age-related” or “ageing-related”? Evidence from meta-analysis of dementia prevalence in the oldest old. Lancet 1995, 346, 931–934. [Google Scholar]
- Bobowick, AR; Brody, JA. Epidemiology of motor-neuron diseases. N. Engl. J. Med 1973, 288, 1047–1055. [Google Scholar]
- Baldereschi, M; Di Carlo, A; Rocca, WA; Vanni, P; Maggi, S; Perissinotto, E; Grigoletto, F; Amaducci, L; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Twofold higher incidence in men. ILSA Working Group. Italian Longitudinal Study on Aging. Neurology 2000, 55, 1358–1363. [Google Scholar]
- Li, TM; Alberman, E; Swash, M. Clinical features and associations of 560 cases of motor neuron disease. J. Neurol. Neurosurg. Psychiatry 1990, 53, 1043–1045. [Google Scholar]
- Brooks, BR. Risk factors in the early diagnosis of ALS: North American epidemiological studies. ALS Care Study Group. Amyotroph. Lateral Scler. Other Motor Neuron Disord 2000, 1, 19–26. [Google Scholar]
- Rowland, LP. Diagnosis of amyotrophic lateral sclerosis. J. Neurol. Sci 1998, 160, 6–24. [Google Scholar]
- Mulder, DW; Howard, FM. Patient resistance and prognosis in amyotrophic lateral sclerosis. MayoClin. Proc 1976, 51, 537–541. [Google Scholar]
- Gubbay, SS; Kahana, E; Zilber, N; Cooper, G; Pintov, S; Leibowitz, Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol 1985, 232, 295–300. [Google Scholar]
- Chen, LC; Smith, A; Ben, Y; Zukic, B; Ignacio, S; Moore, D; Lee, N. Temporal gene expression patterns in G93A/SOD1 mouse. Amyotroph. Lateral. Scler. Other Motor Neuron Disord 2004, 5, 164–171. [Google Scholar]
- Andersen, PM; Sims, KB; Xin, WW; Kiely, R; O’Neill, G; Ravits, J; Pioro, E; Harati, Y; Brower, RD; Levine, JS; Heinicke, HU; Seltzer, W; Boss, M; Brown, RH, Jr. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord 2003, 4, 62–73. [Google Scholar]
- Rothstein, JD; Martin, LJ; Kuncl, RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Eng. J. Med 2002, 326, 1464–1468. [Google Scholar]
- Hayashi, M; Araki, S; Arai, N; Kumada, S; Itoh, M; Tamagawa, K; Oda, M; Morimatsu, Y. Oxidative stress and disturbed glutamate transport in spinal muscular atrophy. Brain Dev 2002, 24, 770–775. [Google Scholar]
- Spreux-Varoquaux, O; Bensimon, G; Lacomblez, L; Salachas, F; Pradat, PF; Le Forestier, N; Marouan, A; Dib, M; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci 2002, 193, 73–78. [Google Scholar]
- Guo, H; Lai, L; Butchbach, MER; Stockinger, MP; Shan, X; Bishop, GA; Lin, CG. Increased expression of the glial glutamate transporter EAAT2 modulates excitotoxicity and delays the onset but not the outcome of ALS in mice. Hum. Mol. Genet 2003, 12, 2519–2532. [Google Scholar]
- Haggstrom, B; Andersen, PM; Hjalmarsson, K; Binzer, M; Forsgren, L. Autoimmunity and ALS: Studies on antibodies to acetylcholinesterase in sera. Acta. Neurol. Scand 1997, 95, 111–114. [Google Scholar]
- Weiss, MD; Weydt, P; Carter, GT. Current pharmacological management of Amyotrophic Lateral Sclerosis and a role for rational polypharmacy. Exp. Opin. Pharmacother 2004, 5, 1231. [Google Scholar]
- Forman, MS; Trojanowski, JQ; Lee, VMY. TDP-43: A novel neurodegenerative proteinopathy. Curr. Opin. Neurobiol 2007, 17, 548–555. [Google Scholar]
- Hasegawa, M; Arai, T; Nonaka, T; Kametani, F; Yoshida, M; Hashizume, Y; Beach, TG; Buratti, E; Baralle, F; Morita, M; Nakano, I; Oda, T; Tsuchiya, K; Akiyama, H. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol 2008, 64, 60–70. [Google Scholar]
- van den Berg, LH; van den Berg, JP; Mathus-Vliegen, EM; Kampelmacher, MJ; van Kesteren, RG; Jennekens, FG. The symptomatic treatment of amyotrophic lateral sclerosis. Ned. Tijdschr. Geneeskd 2004, 148, 513–518. [Google Scholar]
- Miller, RG; Mitchell, JD; Lyon, M; Moore, DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev 2007, 1. No. CD001447. DOI: 10.1002/14651858.CD001447. [Google Scholar]
- MacDonald, ME; Novelletto, A; Lin, C; Tagle, D; Barnes, G; Bates, G; Taylor, S; Allitto, B; Altherr, M; Myers, R; Lehrach, H; Collins, FS; Wasmuth, JJ; Frontali, M; Gusella, JF. The Huntington’s disease candidate region exhibits many different haplotypes. Nat. Genet 1992, 1, 99–103. [Google Scholar]
- Jana, NR; Dikshit, P; Goswami, A; Kotliarova, S; Murata, S; Tanaka, K; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem 2005, 280, 11635–11640. [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- Rubinsztein, DC; Leggo, J; Coles, R; Almqvist, E; Biancalana, V; Cassiman, JJ; Chotai, K; Connarty, M; Crauford, D; Curtis, A; et al. Phenotypic characterization of individuals with 30–40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Genet 1996, 59, 16–22. [Google Scholar]
- Sathasivam, K; Amaechi, I; Mangiarini, L; Bates, GP. Identification of an HD patient with a (CAG)180 repeat expansion and the propagation of highly expanded CAG repeats in lambda phage. Hum. Genet 1997, 99, 692–695. [Google Scholar]
- Lin, B; Nasir, J; MacDonald, H; Hutchinson, G; Graham, RK; Rommesns, JM; Hayden, MR. Sequence of the murine Huntington’s disease gene: evidence for conservation, and polymorphism in a triplet (CCG) repeat alternate splicing. Hum. Mol. Genet 1994, 3, 85–92. [Google Scholar]
- Baxendale, S; Abdulla, S; Elgar, G; Buck, D; Berks, M; Micklem, G; Durbin, R; Bates, G; Brenner, S; Beck, S; Lehrach, H. Comparative sequence analysis of the human and puffer fish Huntington’s disease gene. Nat. Genet 1995, 10, 67–75. [Google Scholar]
- Wang, J; Gines, S; MacDonald, ME; Gusella, JF. Reversal of a full-length mutant huntingtin neuronal cell phenotype by chemical inhibitors of polyglutamine-mediated aggregation. BMC Neurosci 2005, 6, 1. [Google Scholar]
- Trushina, E; McMurray, CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007, 145, 1223–1248. [Google Scholar]
- Myers, RH; Vonsattel, JP; Paskevich, PA; Kiely, DK; Stevens, TJ; Cupples, LA; Richardson, EP, Jr; Bird, ED. Decreased neuronal and increased oligo dendroglial densities in Huntington’s disease caudate nucleus. J. Neuropathol. Exp. Neurol 1991, 50, 729–742. [Google Scholar]
- de la Monte, SM; Vonsattel, JP; Richardson, EP, Jr. Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatumin Huntington’s disease. J. Neuropathol. Exp. Neurol 1988, 47, 516–525. [Google Scholar]
- Sathasivam, K; Amaechi, I; Mangiarini, L; Bates, GP. Identification of an HD patient with a (CAG) 180 repeat expansion and the propagation of highly expanded CAG repeats in lambda phage. Hum Genet 1997, 99, 692–695. [Google Scholar]
- Li, Z; Karlovich, CA; Fish, MP; Scott, MP; Myers, RM. A putative Drosophila homolog of the Huntington’s disease gene. Hum. Mol. Genet 1999, 8, 1807–1815. [Google Scholar]
- Andrade, MA; Bork, P. HEAT repeats in the Huntington’s disease protein [letter]. Nat Genet 1995, 11, 115–116. [Google Scholar]
- Wanker, EE; Rovira, C; Scherzinger, E; Hasenbank, R; Walter, S; Tait, D; Colicelli, J; Lehrach, H. HIP-I: a huntingtin interacting protein isolated by the yeast two-hybrid system. Hum Mol Genet 1997, 6, 487–495. [Google Scholar]
- Holtzman, DA; Yang, S; Drubin, DG. Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that controls membrane cytoskeleton assembly in Saccharomyces erevisiae. J. Cell Biol 1993, 122, 635–644. [Google Scholar]
- Wesp, A; Hicke, L; Palecek, J; Lombardi, R; Aust, T; Munn, AL; Riezman, H. End4p/Sla2p interacts with actin-associated proteins for endocytosis in Saccharomyces cerevisiae. Mol Biol Cell 1997, 8, 2291–2306. [Google Scholar]
- Velier, J; Kim, M; Schwarz, C; Kim, TW; Sapp, E; Chase, K; Aronin, N; DiFiglia, M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol 1998, 152, 34–40. [Google Scholar]
- Engqvist-Goldstein, AE; Kessels, MM; Chopra, VS; Hayden, MR; Drubin, DG. An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of lathrin-coated pits and vesicles. J Cell Biol 1999, 147, 1503–1518. [Google Scholar]
- Li, XJ; Li, SH; Sharp, AH; Nucifora, FC, Jr; Schilling, G; Lanahan, A; Worley, P; Snyder, SH; Ross, CA. A huntingtin-associated protein enriched in brain with implications for pathology. Nature 1995, 378, 398–402. [Google Scholar]
- Block-Galarza, J; Chase, KO; Sapp, E. Fast transport and retrograde movement of huntingtin and HAP 1 in axons. Neuroreport 1997, 8, 2247–2251. [Google Scholar]
- Engelender, S; Sharp, AH; Colomer, V; Tokito, MK; Lanahan, A; Worley, P; Holzbaur, EL; Ross, CA. Huntingtin-associated protein1 (HAP1) interacts with the p150 glued subunit of dynactin. Hum Mol Genet 1997, 6, 2205–2212. [Google Scholar]
- Giorgini, F; Guidetti, P; Nguyen, Q; Bennett, SC; Muchowski, PJ. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nat. Genet 2005, 37, 526–31. [Google Scholar]
- Vonsattel, JP; Myers, RH; Stevens, TJ; Ferrante, RJ; Bird, ED; Richardson, EP. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol 1985, 44, 559–577. [Google Scholar]
- Adam, OR; Jankovic, J. Symptomatic treatment of Huntington’s disease. Neurotherapeutics 2008, 5, 181–197. [Google Scholar]
- Squitieri, F; Cannella, M; Porcellini, A; Brusa, L; Simonelli, M; Ruggieri, S. Short-term effects of olanzapine in Huntington’s disease. Neuropsych Neuropsychol Behav Neurol 2001, 14, 69–72. [Google Scholar]
- Laks, J; Rocha, M; Capitão, C; Domingues, RC; Ladeia, G; Lima, M; Engelhardt, E. Functional and motor response to low dose olanzapine in Huntington’s disease: Case report. Arq. Neuropsiquiatr 2004, 62, 1092–1094. [Google Scholar]
- Grove, VE, Jr; Quintanilla, J; DeVaney, GT. Improvement of Huntington’s disease with olanzapine and valproate. N. Eng. J. Med 2001, 343, 973–974. [Google Scholar]
- Chen, WX; Wang, P; Yan, S; Li, Y; Yu, C; Jiang, L. Acquired hepatocerebral degeneration: A case report. World J. Gastroenterol 2005, 11, 764–766. [Google Scholar]
- Feany, MB; Dickson, DW. Neurodegenerative disorders with extensive tau pathology: A comparative study and review. Ann. Neurol 1996, 40, 139–148. [Google Scholar]
- Umahara, T; Uchihara, T; Tsuchiya, K; Nakamura, A; Ikeda, K; Iwamoto, T; Takasaki, M. Immunolocalization of 14–3-3 isoforms in brains with Pick body disease. Neurosci. Lett 2004, 371, 215–219. [Google Scholar]
- López, C; Comabella, M; Al-Zayat, H; Tintoré, M; Montalban, X. Altered maturation of circulating dendritic cells in primary progressive MS patients. J. Neuroimmunol 2006, 175, 183–191. [Google Scholar]
- Haslam, C. Managing bladder symptoms in people with multiple sclerosis. Nurs. Times 2005, 101, 48–50. [Google Scholar]
- Trapp, BD; Ransohoff, R; Rudick, R. Axonal pathology in multiple sclerosis: relationship to neurologic disability. Curr. Opin. Neurol 1999, 12, 295–302. [Google Scholar]
- Oldstone, MB. Molecular mimicry, microbial infection, and autoimmune disease: evolution of the concept. Curr. Top. Microbiol. Immunol 2005, 296, 1–17. [Google Scholar]
- Schapira, AH. Present and future drug treatment for Parkinson’s disease. J. Neurol. Neurosurg. Psych 2005, 76, 1472–1478. [Google Scholar]
- Lleo, A; Greenberg, SM; Growdon, JH. Current pharmacotherapy for Alzheimer’s disease. Ann. Rev. Med 2006, 57, 513–533. [Google Scholar]
- Gsell, W; Jungkunz, G; Riederer, P. Functional neurochemistry of Alzheimer’s disease. Curr. Pharm. Des 2004, 10, 265–293. [Google Scholar]
- Weiner, MF; Hynan, LS; Parikh, B; Zaki, N; White, CL; Bigio, EH; Lipton, AM; Martin-Cook, K; Svetlik, DA; Cullum, CM; Vobach, S; Rosenberg, RN. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J. Geriatr. Psych. Neurol 2003, 16, 245–250. [Google Scholar]
- Amatniek, JC; Hauser, WA; DelCastillo-Castaneda, C; Jacobs, DM; Marder, K; Bell, K; Albert, M; Brandt, J; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar]
- Del Vecchio, RA; Gold, LH; Novick, SJ; Wong, G; Hyde, LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci. Lett 2004, 367, 164–167. [Google Scholar]
- Mark, RJ; Ashford, JW; Goodman, Y; Mattson, MP. Anticonvulsants attenuate amyloid-peptide neurotoxicity, Ca2+ deregulation, and cytoskeletal pathology. Neurobiol. Aging 1995, 16, 187–198. [Google Scholar]
- Mattson, MP. Pathways towards and away from Alzheimer’s disease. Nat 2004, 430, 631–639. [Google Scholar]
- Palop, JJ; Chin, J; Bien-Ly, N; Massaro, C; Yeung, BZ; Yu, G; Mucke, L. Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J. Neurosci 2005, 25, 9686–9693. [Google Scholar]
- Palop, JJ; Jones, B; Kekonius, L; Chin, J; Yu, G; Raber, J; Masliah, E; Mucke, L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl Acad. Sci 2003, 100, 9572–9577. [Google Scholar]
- DeKosky, ST; Ikonomovic, MD; Styren, SD; Beckett, L; Wisniewski, S; Bennett, DA; Cochran, EJ; Kordower, JH; Mufson, EJ. Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol 2002, 51, 145–155. [Google Scholar]
- Small, DH. Do acetylcholinesterase inhibitors boost synaptic scaling in Alzheimer’s disease? Trends Neurosci 2004, 27, 245–249. [Google Scholar]
- Dickerson, BC; Salat, DH; Greve, DN; Chua, EF; Rand-Giovannetti, E; Rentz, DM; Bertram, L; Mullin, K; Tanzi, RE; Blacker, D; Albert, MS; Sperling, RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005, 65, 404–411. [Google Scholar]
- Yang, Q; Williams, D; Owusu-Ababio, G; Ebube, NK; Habib, MJ. Controlled release tacrine delivery system for the treatment of Alzheimer’s disease. Drug Deliv 2001, 8, 93–98. [Google Scholar]
- Vinogradov, SV; Batrakova, EV; Kabanov, AV. Nanogels for oligonucleotide delivery to the brain. Bioconjug. Chem 2004, 15, 50–60. [Google Scholar]
- Rice, A; Michaelis, ML; Georg, G; Liu, Y; Turunen, B; Audus, KL. Overcoming the blood-brain barrier to taxane delivery for neurodegenerative diseases and brain tumors. J. Mol. Neurosci 2003, 20, 339–343. [Google Scholar]
- Uzan, G. Therapeutic use of stem cells. II. Adult stem cells. Rev. Prat 2004, 54, 1515–1527. [Google Scholar]
- Shoichet, M. Shoichet lab, McEwen Centre for Regenerative Medicine.
- Vernalis (Pty) Ltd. V1512-Treatment of Parkinson’s disease.
- Morgan, JC; Sethi, KD. Rotigotine for the treatment of Parkinson’s disease. Exp. Rev. Neurother 2006, 6, 1275–1282. [Google Scholar]
- Guldenpfennig, WM; Poole, KH; Sommerville, KW; Boroojerdi, B. Safety, tolerability, and efficacy of continuous transdermal dopaminergic stimulation with rotigotine patch in early-stage idiopathic Parkinson disease. Clin. Neuropharmacol 2005, 28, 106–110. [Google Scholar]
- Watts, RL; Jankovic, J; Waters, C; Rajput, A; Boroojerdi, B; Rao, J. Randomized, blind, controlled trial of transdermal rotigotine in early Parkinson disease. Neurology 2007, 68, 272–276. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
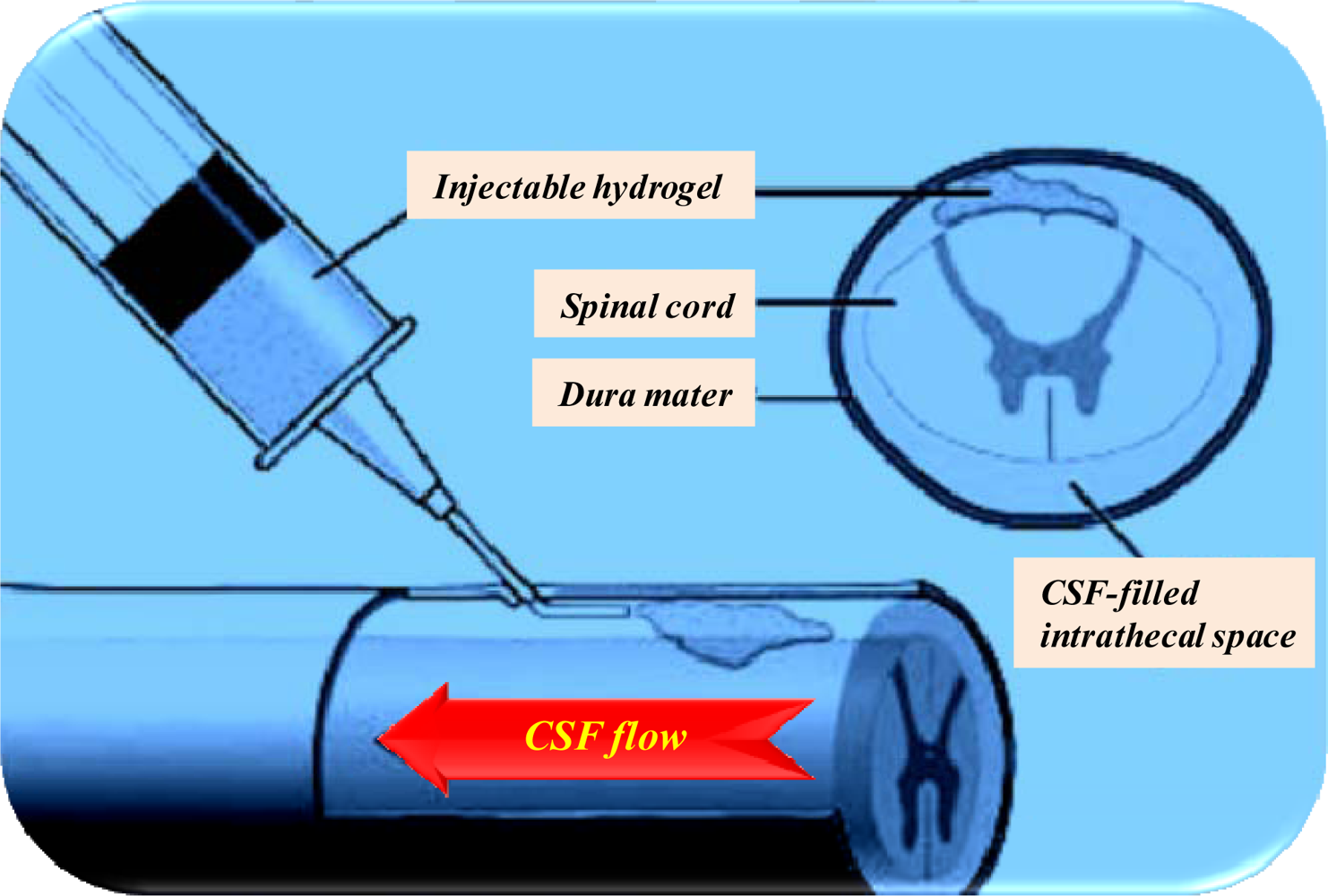{kind=link}
| Class of Drug | Therapeutic/Pharmacological Activity |
|---|---|
| Protease inhibitors | Decrease activity of β- and γ-secretase that cleave Aβ from APP |
| Extracellular Aβ-binding compounds e.g. Cu2+, Zn2+ chelators | Prevent aggregation of Aβ into cytotoxic amyloid fibrils |
| Immunotherapeutic agents | Induces local and T-cell innate immune response. |
| Non-steroidal anti-inflammatory drugs, e.g. naproxen, celecoxib, aspirin | Dampens the innate immune-response thereby delaying the progression of AD |
| Neuroprotective agents e.g. antioxidants, MAO-inhibitors, Ca-channel blockers and anti-apoptotics | Interferes with the mechanisms of Aβ-triggered putative neurotoxicity |
| Statins, e.g. simvastatin | Decreases cholesterol which is a major risk factor in amyloid accumulation thereby lowering the risk of AD |
| Hormonal replacement, e.g. estrogens | Decreases the risk of developing AD in postmenopausal woman. |
| Cholinergic replacement agents and cholinesterase inhibitors, e.g. tacrine, donepezil, galantamine | Symptomatic treatment of AD |
| Trophic factors | Prevent degeneration of axotomized cholinergic septal and BF neurons by regulating hippocampal NGF, APP and ACh-mediated activity |
| Environmental enrichment agents | Lead to pronounced reductions in cerebral amyloid deposits thereby lowering the risk of AD |
| Features | Atypical Parkinsonianism | Typical Parkinsonianism |
|---|---|---|
| Pathological hallmark | Loss of substantia nigra cells and neuronal cell degeneration containing DA receptors in parts of the CNS such as striatum as well as Dementia with Lewy bodies (DLB) cortico-basal ganglionic degeneration (CBD) | Loss of substantia nigra cells, preserved cells in striatum (basal ganglia) and response to DA stimulation. |
| Clinical symptoms | Resting tremor, slowed movement, muscle rigidity, postural instability as well as vertical gaze palsy, early postural instability (progressive supranuclear palsy) and multiple system atrophy (anterocollis). | Resting tremor, slowed movement, muscular rigidity, postural instability. |
| Heredity and familyhistory | More sporadic than familial. | Sporadic and familial. Family history plays a major role. |
| Genetic involvement | Tau positive NFTs present as neuronal inclusions, no Lewy bodies. | Lewy bodies, mutation of α-synuclein, parkin and ubiquitin genes. |
| Aetiology | Sporadic, toxins, MPTP, viruses. | Mostly genetic, mitochondrial damage, cell protein disposal. |
| Inflammation | Inflammation of substantia nigra and basal ganglia. | Inflammation of the basal ganglia. |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Choonara, Y.E.; Pillay, V.; Du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.K.; Sibambo, S.R. Trends in the Molecular Pathogenesis and Clinical Therapeutics of Common Neurodegenerative Disorders. Int. J. Mol. Sci. 2009, 10, 2510-2557. https://doi.org/10.3390/ijms10062510
Choonara YE, Pillay V, Du Toit LC, Modi G, Naidoo D, Ndesendo VMK, Sibambo SR. Trends in the Molecular Pathogenesis and Clinical Therapeutics of Common Neurodegenerative Disorders. International Journal of Molecular Sciences. 2009; 10(6):2510-2557. https://doi.org/10.3390/ijms10062510
Chicago/Turabian StyleChoonara, Yahya E., Viness Pillay, Lisa C. Du Toit, Girish Modi, Dinesh Naidoo, Valence M.K. Ndesendo, and Sibongile R. Sibambo. 2009. "Trends in the Molecular Pathogenesis and Clinical Therapeutics of Common Neurodegenerative Disorders" International Journal of Molecular Sciences 10, no. 6: 2510-2557. https://doi.org/10.3390/ijms10062510