Classification of 5-HT1A Receptor Ligands on the Basis of Their Binding Affinities by Using PSO-Adaboost-SVM

Abstract
:1. Introduction
2. Results and Discussion
2.1. Variable selection and model building
2.1.1. Variable selection by stepwise-MLR
2.1.2. Variable selection by PSO
2.2. Comparison with different models
2.3. Variable’s interpretation of the best model
2.4. Comparison with other approaches
3. Experimental and the Theory of the Modeling Methods
3.1. Experimental
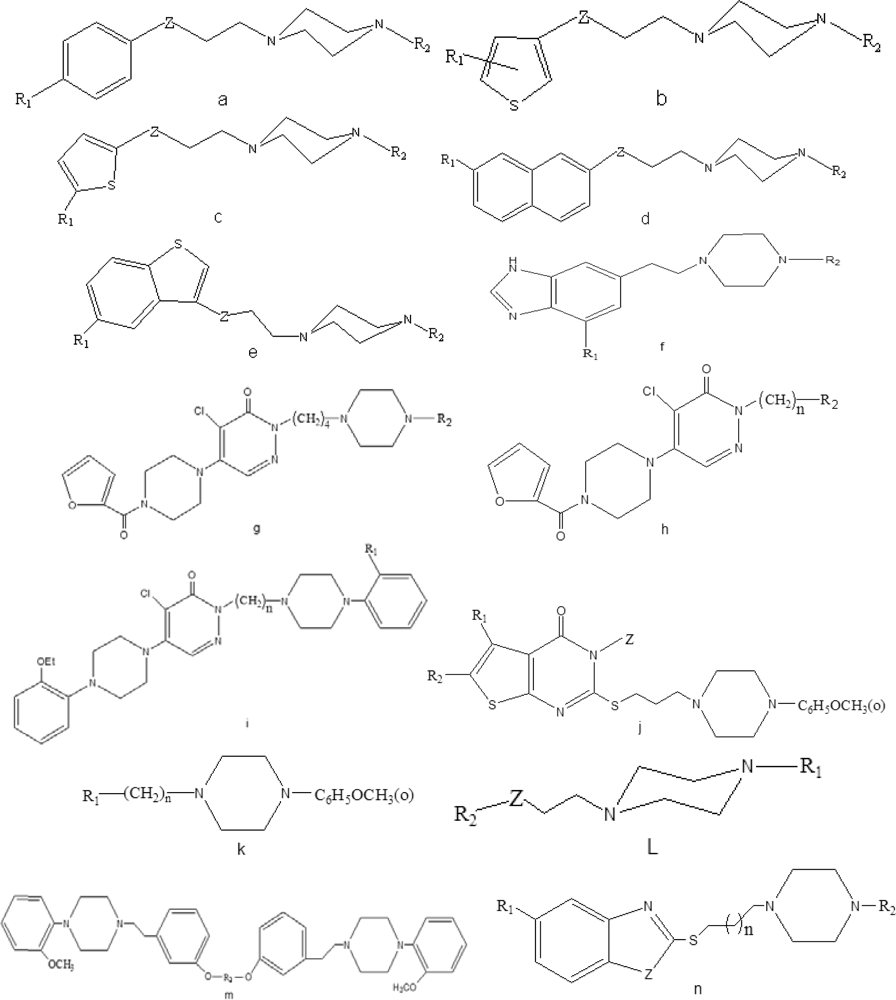
3.1.1. Data sets
3.1.2. Descriptor calculation
3.2. The theory of the modeling methods
3.2.1. Theory of PSO
3.2.2. Theory of AdaBoost algorithm
- Step 1 Distribution D over the N training examples, initializes the weight vector:
- Step 2 Do for t = 1, 2,…, T
3.2.3. Methodology
3.2.4. The evaluation of prediction power
4. Conclusions
References and Notes
- Siracusa, MA; Salerno, L; Modica, MN; Pittalá, V; Romeo, G; Amato, ME; Nowak, M; Bojarski, AJ; Mereghetti, I; Cagnotto, A; Mennini, T. Synthesis of new arylpiperazinylalkylthiobenzimidazole, benzothiazole, or benzoxazole derivatives as potent and selective 5-HT1A serotonin receptor ligands. J. Med. Chem 2008, 51, 4529–4538. [Google Scholar]
- Uphouse, L. Multiple serotonin receptors: Too many, not enough, or just the right number? Neurosci. Biobehav. Rev 1997, 21, 679–698. [Google Scholar]
- Abbas, SY; Nogueira, MI; Azmitia, EC. Antagonist-induced increase in 5-HT1A-receptor expression in adult rat hippocampus and cortex. Synapse 2007, 61, 531–539. [Google Scholar]
- Ögren, SO; Eriksson, TM; Elvander-Tottie, E; D’Addario, C; Ekström, JC; Svenningsson, P; Meister, B; Kehr, J; Stiedl, O. The role of 5-HT1A receptors in learning and memory. Behav. Brain Res 2008, 195, 54–77. [Google Scholar]
- Millan, MJ; Gobert, A; Roux, S; Porsolt, R; Meneses, A; Carli, M. The serotonin1A receptor partial agonist S15535 [4-(benzodioxan-5-yl)1-(indan-2-yl)piperazine] enhances cholinergic transmission and cognitive function in rodents: a combined neurochemical and behavioral analysis. J. Pharmacol. Exp. Ther 2004, 311, 190–203. [Google Scholar]
- Bert, B; Voigt, JP; Kusserow, H; Theuring, F; Rex, A; Fink, H. Increasing the number of 5-HT1A-receptors in cortex and hippocampus does not induce mnemonic deficits in mice. Pharmacol. Biochem. Behav 2009, 92, 76–81. [Google Scholar]
- Hjorth, S; Sharp, T. Effect of the 5-HT1A receptor agonist 8-OH-DPAT on the release of 5-HT in dorsal and median raphe-innervated rat brain regions as measured by in vivo microdialysis. Life Sci 1991, 48, 1779–1786. [Google Scholar]
- Belcheva, I; Belcheva, S; Petkov, VV; Hadjiivanova, C; Petkov, VD. Behavorial responses to the 5-HT1A receptor antagonist NAN190 injected into rat CA1 hippocampal area. Gen. Pharmacol 1997, 28, 435–441. [Google Scholar]
- Galeotti, N; Ghelardini, C; Bartolini, A. Role of 5-HT1A receptors in a mouse passive avoidance paradigm. Jpn. J. Pharmacol 2000, 84, 418–424. [Google Scholar]
- Misane, I; Ögren, SO. Selective 5-HT1A antagonistsWAY 100635 and NAD-299 attenuate the impairment of passive avoidance caused by scopolamine in the rat. Neuropsychopharmacology 2003, 28, 253–264. [Google Scholar]
- Meneses, A; Perez-Garcia, G. 5-HT(1A) receptors and memory. Neurosci. Biobehav. Rev 2007, 31, 705–727. [Google Scholar]
- Negus, SS. Some implications of receptor theory for in vivo assessment of agonists, antagonists and inverse agonists. Biochem. Pharmacol 2006, 71, 1663–1670. [Google Scholar]
- Weber, KC; da Silva, ABF. A chemometric study of the 5-HT1A receptor affinities presented by arylpiperazine compounds. Eur. J. Med. Chem 2008, 43, 364–372. [Google Scholar]
- Andrić, D; Roglić, G; Šukalović, V; Šoškić, V; Rajačić, SK. Synthesis binding properties and receptor docking of 4-halo-6-[2-(4-arylpiperazin-1-yl) ethyl]-1H-benzimidazoles, mixed ligands of D2 and 5-HT1A receptors. Eur. J. Med. Chem 2008, 43, 1696–1705. [Google Scholar]
- Betti, L; Zanelli, M; Giannaccini, G; Manetti, F; Schenoned, S; Strappaghetti, G. Synthesis of new piperazine–pyridazinone derivatives and their binding affinity toward α1-, α2-adrenergic and 5-HT1A serotoninergic receptors. Bioorgan. Med. Chem 2006, 14, 2828–2836. [Google Scholar]
- Modica, M; Santagati, M; Russo, F; Selvaggini, C; Cagnotto, A; Mennini, T. High affinity and selectivity of [[(arylpiperazinyl) alkyl]thio]thieno[2,3-d]pyrimidinone derivatives for the 5-HT1A receptor Synthesis and structure–affinity relationships. Eur. J. Med. Chem 2000, 35, 677–689. [Google Scholar]
- Oficialdegui, AM; Martinez, J; Pérez, S; Heras, B; Irurzun, M; Palop, JA; Tordera, R; Lasheras, B; del Río, J; Monge, A. Design, synthesis and biological evaluation of new 3-[(4-aryl) piperazin-1-yl]-1-arylpropane derivatives as potential antidepressants with a dual mode of action: Serotonin reuptake inhibition and 5-HT1A receptor antagonism. Farmaco Prat 2000, 55, 345–353. [Google Scholar]
- Leopoldo, M; Lacivita, E; Colabufo, NA; Niso, M; Berardi, F; Perrone, R. Bivalent ligand approach on 4-[2-(3-methoxyphenyl) ethyl]-1-(2-methoxyphenyl)piperazine: Syntheis and binding affinities for 5-HT7 and 5-HT1A receptors. Bioorgan. Med. Chem 2007, 15, 5316–5321. [Google Scholar]
- Putz, MV; Lacrămă, AM. Introducing spectral structure activity relationship (s-sar) analysis. Application to ecotoxicology. Int. J. Mol. Sci 2007, 8, 363–391. [Google Scholar]
- Putz, MV; Putz, AM; Lazea, M; Ienciu, L; Chiriac, A. Quantum-SAR extension of the Spectral-SAR lgorithm. Application to polyphenolic anticancer bioactivity. Int. J. Mol. Sci 2009, 10, 1193–1214. [Google Scholar]
- Chilmonczyk, Z; Leś, A; Woźniakowska, A; Cybulski, J; Kozioł, AE; Gdaniec, M. Buspirone analogues as ligands of the 5-HT1A receptor. 1. The molecular structure of buspirone and its two analogues. J. Med. Chem 1995, 38, 1701–1710. [Google Scholar]
- Borosy, AP; Morvay, M; Mátyus, P. 3D QSAR analysis of novel 5-HT receptor ligands. Chemom. Intell. Lab Syst 1999, 47, 239–252. [Google Scholar]
- Maciejewska, D; Żołek, T; Herold, F. CoMFA methodology in structure-activity analysis of hexahydro- and octahydropyrido[1,2-c]pyrimidine derivatives based on affinity towards 5-HT1A, 5-HT2A and a1-adrenergic receptors. J. Mol. Graph. Model 2006, 25, 353–362. [Google Scholar]
- Guccione, S; Doweyko, AM; Chen, HM; Barrettad, GU; Balzano, F. 3D-QSAR using ‘Multiconformer’ alignment: The use of HASL in the analysis of 5-HT1A thienopyrimidinone ligands. J. Comput. Aided Mol. Des 2000, 14, 647–657. [Google Scholar]
- Debnath, B; Samanta, S; Naska, SK; Roy, K; Jha, T. QSAR study on the affinity of some arylpiperazines towards the 5-HT1A/α1-adrenergic receptor using the E-state index. Bioorg. Med. Chem. Lett 2003, 13, 2837–2842. [Google Scholar]
- Kuźmin, VE; Artemenko, AG; Polischuk, PG; Muratov, EN; Hromov, AI; Liahovskiy, AV; Andronati, SA; Makan, SY. Hierarchic system of QSAR models (1D-4D) on the base of simplex representation of molecular structure. J. Mol. Model 2005, 11, 457–467. [Google Scholar]
- Ghasemi, JB; Abdolmaleki, A; Mandoumi, N. A quantitative structure property relationship for prediction of solubilization of hazardous compounds using GA-based MLR in CTAB micellar media. J. Hazard. Mater 2009, 161, 74–80. [Google Scholar]
- Clementi, S; Wold, S. How to choose the proper statistical method. In Chemometric methods in molecular design; van de Waterbeemd, H, Ed.; VCH: New York, USA, 1995; pp. 319–338. [Google Scholar]
- Heravi, MJ; Baboli, MA; Shahbazikhah, P. QSAR study of heparanase inhibitors activity using artificial neural networks and LevenbergeMarquardt algorithm. Eur. J. Med. Chem 2008, 43, 548–556. [Google Scholar]
- Haus, F; Boissel, O; Junter, GA. Multiple regression modelling of mineral base oil biodegradability based on their physical properties and overall chemical composition. Chemosphere 2003, 50, 939–948. [Google Scholar]
- Randić, M. On characterization of cyclic structures. J. Chem. Inf. Comput. Sci 1997, 37, 1063–1071. [Google Scholar]
- Balaban, AT. Five new topological indices for the branching of tree-like graphs. Theor. Chim. Acta 1979, 53, 355–375. [Google Scholar]
- Hu, QN; Liang, YZ; Ren, FL. Molecular graph center, a novel approach to locate the center of a molecule and a new centric index. J. Mol. Struc.-Theochem 2003, 635, 105–113. [Google Scholar]
- Bonchev, D. Ovellrall connectivity and topological complexity: A new tool for QSPR/QSAR. In Topological indices and related descriptors in QSAR and QSPR; Deviller, J, Balaban, AT, Eds.; Gordon & Breach Science Publishers: Amsterdam, The Netherlands, 1999. [Google Scholar]
- Gramatica, P; Corradi, M; Consonni, V. Modelling and prediction of soil sorption coefficients of non-ionic organic pesticides by molecular descriptors. Chemosphere 2000, 41, 763–777. [Google Scholar]
- Kier, LB; Hall, LH; Frazer, JW. An index of electrotopological state for atoms in molecules. J. Math. Chem 1991, 7, 229–241. [Google Scholar]
- Konstantinova, EV. The discrimination ability of some topological and information distance indices for graphs of unbranched hexagonal systems. J. Chem. Inf. Comput. Sci 1996, 36, 54–57. [Google Scholar]
- Chari, R; Qureshi, F; Moschera, J; Tarantino, R; Kalonia, D. Development of improved empirical models for estimating the binding constant of a β-cyclodextrin inclusion complex. Pharm Res 2009, 26. (in press).. [Google Scholar]
- Xu, J; Liu, L; Xu, WL; Zhao, SP; Zuo, DY. A general QSPR model for the prediction of θ (lower critical solution temperature) in polymer solutions with topological indices. J. Mol. Graph. Model 2007, 26, 352–359. [Google Scholar]
- Todeschini, R; Consonni, V. Handbook of molecular descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Cheng, YC; Prussof, WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (IC50) of a enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [Google Scholar]
- MolinspirationWebME Editor 2.6. http://www.molinspiration.com:9080/mi/webme.html (accessed 4 January, 2009).
- Virtual Computational Chemistry Laboratory. http://www.vcclab.org/lab/edragon/ (accessed 9 January, 2009).
- Li, XM; Zhao, M; Tang, YR; Wang, C; Zhang, ZD; Peng, SQ. N-[2-(5,5-Dimethyl-1,3-dioxane-2-yl)ethyl]amino acids: Their synthesis, anti-inflammatory evaluation and QSAR analysis. Eur. J. Med. Chem 2008, 43, 8–18. [Google Scholar]
- Sabet, R; Fassihi, A. QSAR study of antimicrobial 3-hydroxypyridine-4-one and 3-hydroxypyran-4-one derivatives using different chemometric tools. Int. J. Mol. Sci 2008, 9, 2407–2423. [Google Scholar]
- Liu, JW; Wu, GF; Cui, GH; Wang, WX; Zhao, M; Wang, C; Zhang, ZD; Peng, SQ. A new class of anti-thrombosis hexahydropyrazino-[1’,2’:1,6] pyrido-[3,4-b]-indole-1,4-dions: Design, synthesis, logK determination, and QSAR analysis. Bioorgan. Med. Chem 2007, 15, 5672–5693. [Google Scholar]
- Kennedy, J; Eberhart, RC. Particle swarm optimization. Proceedings of the IEEE International Joint Conference on Neural Networks, Perth, Australia, November 27–December 1, 1995; pp. 1942–1948.
- Kennedy, J; Eberhart, RC. Swarm Intelligence; Morgan Kaufmann Publishers: San Francisco, CA, USA, 2001. [Google Scholar]
- Shi, YH; Eberhart, R. A Modified Particle Swarm Optimizer. Proceedings of the IEEE International Conference on Evolutionary Computation, Anchorage, USA, May 4–9, 1998; pp. 69–73.
- Mandal, KK; Basu, M; Chakraborty, N. Particle swarm optimization technique based short-term hydrothermal scheduling. Appl. Soft Comput 2008, 8, 1392–1399. [Google Scholar]
- Lee, SH; Kim, HI; Cho, NI; Jeong, YH; Chung, KS; Jun, CS. Automatic Defect Classification Using Boosting. IEEE Proceedings of the Fourth International Conference on Machine Learning and Applications (ICMLA’05), Los Angeles, USA, December 15–17, 2005.
- Freund, Y; Schapire, RE. A decision-theoretic generalization of online learning and an application to boosting. J. Comput. System Sci 1997, 55, 119–139. [Google Scholar]
- Friedman, J; Hastie, T; Tibshirani, R. Additive Logistic Regression: a Statistical View of Boosting. Ann. Stat 2000, 28, 337–407. [Google Scholar]
- Zhang, MH; Xu, QS; Daeyaert, F; Lewi, PJ; Massart, DL. Application of boosting to classification problems in chemometrics. Anal. Chim. Acta 2005, 544, 167–176. [Google Scholar]
- Chihchung, C; Chihjen, L. LIBSVM-A Library for Support Vector Machines. http://www.csie.ntu.edu.tw/~cjlin/libsvm (accessed 15 January, 2009).
- Cortes, C; Vapnik, V. Support-vector networks. Mach. Learn 1995, 20, 273–297. [Google Scholar]
- Liu, HX; Zhang, RS; Yao, XJ; Liu, MC; Hu1, ZD; Fan, BT. QSAR and classification models of a novel series of COX-2 selective inhibitors: 1, 5-diarylimidazoles based on support vector machines. J. Comput. Aided Mol. Des 2004, 18, 389–399. [Google Scholar]


{kind=link}
| Symbols | Descriptor definition |
|---|---|
| MSD | mean square distance index (Balaban) |
| MAXDN | maximal electrotopological negative variation |
| MAXDP | maximal electrotopological positive variation |
| PW4 | path/walk 4 - Randic shape index |
| PW5 | path/walk 5 - Randic shape index |
| PJI2 | 2D Petitjean shape index |
| BAC | Balaban centric index |
| ICR | radial centric information index |
| TI1 | first Mohar index TI1 |
| D/D | distance/detour index |
| DELS | molecular electrotopological variation |
| S1K | 1-path Kier alpha-modified shape index |
| ECC | eccentricity |
| MDDD | mean distance degree deviation |
| No. | R1 | R2 | Z | n | pKi | Class
| ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | MS | MAS | PS | PAS | ||||||
| a1 | H | 2-Methoxyphenyl | CHOH | – | 7.32 | 1 | 1 | 1 | 1 | 1 |
| a2 | H | 2-Methoxyphenyl | CHO-4-CF3C6H4 | – | 6.35 | −1 | −1 | −1 | −1 | −1 |
| a3* | H | 2-Methoxyphenyl | CNOH | – | 7.76 | 1 | −1** | 1 | 1 | 1 |
| a4 | H | 4-Chlorophenyl | CO | – | 6.10 | −1 | −1 | −1 | −1 | −1 |
| a5* | H | 4-Chlorophenyl | CHO-4-CH3C6H4 | – | 5.84 | −1 | −1 | −1 | −1 | −1 |
| a6 | H | 4-Chlorophenyl | CHO-3,4-OCH2OC6H3 | – | 6.26 | −1 | −1 | −1 | −1 | −1 |
| a7 | H | 4-Methoxyphenyl | CO | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a8 | H | 4-Methoxyphenyl | CHOH | – | 5.30 | −1 | −1 | −1 | −1 | |
| a9* | H | 2-Chlorophenyl | CO | – | 6.74 | 1 | −1** | −1** | 1 | 1 |
| a10* | H | 2-Chlorophenyl | CHOH | – | 6.94 | 1 | −1** | −1** | 1 | 1 |
| a11 | H | 2-Chlorophenyl | CHO-4-CF3C6H4 | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a12 | H | 4-Fluorophenyl | CO | – | 6.10 | −1 | −1 | −1 | −1 | −1 |
| a13 | H | 4-Fluorophenyl | CHO-4-CF3C6H4 | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a14 | H | 2-Pyridyl | CO | – | 7.30 | 1 | 1 | 1 | 1 | 1 |
| a15* | H | 2-Pyridyl | CHOH | – | 6.81 | 1 | 1 | 1 | 1 | 1 |
| a16 | H | 4-Nitrophenyl | CO | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a17 | H | 4-Nitrophenyl | CHOH | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a18 | H | 4-Nitrophenyl | CHO-4-CF3C6H4 | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a19 | Phenyl | 2-Methoxyphenyl | CO | – | 5.44 | −1 | −1 | −1 | −1 | −1 |
| a20* | Phenyl | 2-Methoxyphenyl | CHO-4-CF3C6H4 | – | 5.30 | −1 | −1 | −1 | 1** | −1 |
| a21 | Methoxy | 2-Methoxyphenyl | CO | – | 5.76 | −1 | −1 | −1 | −1 | −1 |
| a22 | Methoxy | 2-Methoxyphenyl | CHOH | – | 6.49 | −1 | −1 | −1 | −1 | −1 |
| a23 | Methoxy | 2-Methoxyphenyl | CHO-4-CF3C6H4 | – | 6.00 | −1 | −1 | −1 | −1 | −1 |
| a24 | H | 2-Methoxyphenyl | CO | – | 7.30 | 1 | 1 | 1 | 1 | 1 |
| a25 | H | 4-Chlorophenyl | CHOH | – | 6.10 | −1 | −1 | −1 | −1 | −1 |
| a26 | H | 4-Methoxyphenyl | CHO-4-CF3C6H4 | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| a27 | H | 2-Pyrimidyl | CO | – | 6.92 | 1 | 1 | 1 | 1 | 1 |
| a28 | H | 2-Pyrimidyl | CHO-4-CF3C6H4 | – | 5.80 | −1 | −1 | −1 | −1 | −1 |
| a29 | H | 2-Pyridyl | CHO-4-CF3C6H4 | – | 5.80 | −1 | −1 | −1 | −1 | −1 |
| a30 | Phenyl | 2-Methoxyphenyl | CHOH | – | 6.07 | −1 | −1 | −1 | −1 | −1 |
| b1 | H | 2-Methoxyphenyl | CHOH | – | 7.30 | 1 | 1 | 1 | 1 | 1 |
| b2* | H | 2-Methoxyphenyl | CHO-4-CF3C6H4 | – | 6.59 | −1 | −1 | −1 | −1 | −1 |
| b3 | H | 2-Methoxyphenyl | CNOH | – | 8.19 | 1 | 1 | 1 | 1 | 1 |
| b4* | H | 4-Chlorophenyl | CO | – | 6.15 | −1 | −1 | −1 | −1 | −1 |
| b5 | H | 2-Chlorophenyl | CO | – | 6.70 | −1 | −1 | −1 | −1 | −1 |
| b6 | H | 2-Chlorophenyl | CHOH | – | 6.70 | −1 | −1 | −1 | −1 | −1 |
| b7 | H | 1-Naphthyl | CO | – | 7.46 | 1 | 1 | 1 | 1 | 1 |
| b8* | 2,5-Dimethyl | 2-Methoxyphenyl | CO | – | 8.30 | 1 | 1 | 1 | 1 | 1 |
| b9* | 2,5-Dimethyl | 2-Hydroxyphenyl | CO | – | 8.12 | 1 | −1** | 1 | 1 | 1 |
| b10 | 2,5-Dimethyl | 2-Hydroxyphenyl | CHOH | – | 7.05 | 1 | 1 | 1 | 1 | 1 |
| b11 | 2,5-Dimethyl | 1-Naphthyl | CO | – | 7.00 | 1 | 1 | 1 | 1 | 1 |
| b12* | H | H | CO | – | 7.80 | 1 | 1 | 1 | 1 | 1 |
| b13 | H | H | CHOH | – | 5.56 | −1 | −1 | −1 | −1 | −1 |
| b14 | 2,5-Dimethyl | 2,5-Dimethyl | CHOH | – | 7.92 | 1 | 1 | 1 | 1 | 1 |
| c1 | H | 2-Methoxyphenyl | CO | – | 8.00 | 1 | 1 | 1 | 1 | 1 |
| c2 | H | 2-Methoxyphenyl | CHOH | – | 7.72 | 1 | 1 | 1 | 1 | 1 |
| c3 | H | 4-Chlorophenyl | CO | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| c4 | H | 4-Chlorophenyl | CHOH | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| c5 | 5-Methyl | 2-Methoxyphenyl | CO | – | 7.76 | 1 | 1 | 1 | 1 | 1 |
| c6 | 5-Methyl | 2-Methoxyphenyl | CHOH | – | 7.47 | 1 | 1 | 1 | 1 | 1 |
| c7 | 5-Nitro | 2-Methoxyphenyl | CO | – | 6.47 | −1 | −1 | −1 | −1 | −1 |
| d1 | H | 2-Methoxyphenyl | CHOH | – | 6.38 | −1 | −1 | −1 | −1 | −1 |
| d2 | H | 4-Chlorophenyl | CO | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| d3* | H | 4-Chlorophenyl | CHOH | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| d4* | H | 2-Methoxyphenyl | CO | – | 6.60 | −1 | −1 | −1 | −1 | −1 |
| e1 | H | 2-Methoxyphenyl | CO | – | 7.36 | 1 | 1 | 1 | 1 | 1 |
| e2 | H | 2-Methoxyphenyl | CHOH | – | 7.70 | 1 | 1 | 1 | 1 | 1 |
| e3 | H | 4-Chlorophenyl | CO | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| e4 | H | 4-Chlorophenyl | CHOH | – | 5.30 | −1 | −1 | −1 | −1 | −1 |
| e5* | H | 2-Hydroxyphenyl | CO | – | 6.96 | 1 | −1** | 1 | 1 | 1 |
| e6 | H | 2-Hydroxyphenyl | CHOH | – | 7.74 | 1 | 1 | 1 | 1 | 1 |
| e7 | H | 4-Chloro-2-methoxyphenyl | CO | – | 6.30 | −1 | −1 | −1 | −1 | −1 |
| e8* | H | 4-Chloro-2-methoxyphenyl | CHOH | – | 6.44 | −1 | 1** | 1** | −1 | −1 |
| e9 | H | 4-Fluoro-2-methoxyphenyl | CHOH | – | 6.30 | −1 | −1 | −1 | −1 | −1 |
| e10* | H | 1-Naphthyl | CO | – | 7.00 | 1 | 1 | 1 | 1 | 1 |
| e11* | H | 4-Fluoro-2-methoxyphenyl | CO | – | 6.30 | −1 | −1 | −1 | −1 | −1 |
| f1 | Cl | Phe | – | – | 7.05 | 1 | −1** | 1 | 1 | 1 |
| f2 | Cl | 2-MeOPhe | – | – | 8.28 | 1 | 1 | 1 | 1 | 1 |
| f3 | Cl | 2-ClPhe | – | – | 7.34 | 1 | 1 | 1 | 1 | 1 |
| f4 | Cl | 2-CF3Phe | – | – | 7.68 | 1 | 1 | 1 | 1 | 1 |
| f5 | Cl | Pyrimidin-2-yl | – | – | 6.28 | −1 | −1 | 1** | −1 | −1 |
| f6 | Br | Phe | – | – | 7.62 | 1 | 1 | 1 | 1 | 1 |
| f7 | Br | 2-MeOPhe | – | – | 8.85 | 1 | 1 | 1 | 1 | 1 |
| f8* | Br | 2-ClPhe | – | – | 9.85 | 1 | 1 | 1 | 1 | 1 |
| f9 | Br | 2-CF3Phe | – | – | 7.99 | 1 | 1 | 1 | 1 | 1 |
| f10 | Br | Pyrimidin-2-yl | – | – | 6.89 | 1 | 1 | 1 | 1 | 1 |
| f11 | – | Phe | – | – | 6.71 | 1 | 1 | 1 | 1 | 1 |
| f12 | – | 2-MeOPhe | – | – | 7.69 | 1 | 1 | 1 | 1 | 1 |
| f13 | – | 2-ClPhe | – | – | 6.61 | −1 | −1 | −1 | 1** | −1 |
| f14 | – | 2-CF3Phe | – | – | 7.48 | 1 | 1 | 1 | 1 | 1 |
| g1* | – | 2-isopropoxyphenyl | – | – | 8.89 | 1 | 1 | 1 | 1 | 1 |
| g1* | – | 2-ethoxyphenyl | – | – | 7.96 | 1 | 1 | 1 | 1 | 1 |
| g1 | – | 2,3-dihydrobenzo [1,4]dioxin-2-yl-methyl | – | – | 6.63 | −1 | −1 | −1 | −1 | −1 |
| g1 | – | 2-pyrimidyl | – | – | 6.68 | −1 | −1 | −1 | −1 | −1 |
| g1 | – | 3-chlorophenyl | – | – | 7.77 | 1 | 1 | 1 | 1 | 1 |
| g1 | – | 3-trifluoromethylphenyl | – | – | 6.86 | 1 | 1 | 1 | 1 | 1 |
| h1 | – | 4-(3-chlorophenyl)piperazin-1-yl | – | 3 | 6.21 | −1 | −1 | −1 | −1 | −1 |
| h2 | – | 4-(3-trifluorophenyl)piperazin-1-yl | – | 3 | 6.93 | 1 | 1 | 1 | 1 | 1 |
| h3 | – | 4-pyridin-2-yl-piperazin-1-yl | – | 3 | 5.94 | −1 | −1 | −1 | −1 | −1 |
| h4 | – | 4-(2-isopropoxyphenyl)piperazin-1-yl | – | 2 | 8.70 | 1 | 1 | 1 | 1 | 1 |
| i1* | OEt | – | – | 4 | 8.30 | 1 | 1 | 1 | 1 | 1 |
| i2 | OEt | – | – | 7 | 8.49 | 1 | 1 | 1 | 1 | 1 |
| i3 | OiPr | – | – | 4 | 8.41 | 1 | 1 | 1 | 1 | 1 |
| i4 | OiPr | – | – | 7 | 8.14 | 1 | 1 | 1 | 1 | 1 |
| j1* | Me | Me | NH2 | – | 9.80 | 1 | 1 | 1 | 1 | 1 |
| j2 | Me | COOC2H5 | NH2 | – | 8.87 | 1 | 1 | 1 | 1 | 1 |
| j3 | Me | C2H5 | NH2 | – | 9.72 | 1 | 1 | 1 | 1 | 1 |
| j4* | H | Me | C2H5 | – | 8.48 | 1 | 1 | 1 | 1 | 1 |
| j5 | Me | Me | C2H5CH= CH2 | – | 7.90 | 1 | 1 | 1 | 1 | 1 |
| j6 | Me | Me | NHCOCH3 | – | 8.36 | 1 | 1 | 1 | 1 | 1 |
| j7* | Me | Me | H | – | 7.88 | 1 | 1 | 1 | 1 | 1 |
| j8 | Me | Me | Me | – | 8.79 | 1 | 1 | 1 | 1 | 1 |
| j9* | Me | Me | NHC6H5 | – | 6.57 | −1 | −1 | −1 | −1 | −1 |
| k1 | – |  | – | 3 | 9.42 | 1 | 1 | 1 | 1 | 1 |
| k2* | – |  | – | 3 | 9.09 | 1 | 1 | 1 | 1 | 1 |
| k3 | – |  | – | 3 | 8.62 | 1 | 1 | 1 | 1 | 1 |
| k4* | – |  | – | 3 | 8.43 | 1 | 1 | −1** | 1 | 1 |
| k5 | – |  | – | 1 | 5.82 | −1 | −1 | −1 | −1 | −1 |
| k6 | – |  | – | 3 | 8.43 | 1 | 1 | 1 | 1 | 1 |
| k7 | – |  | – | 2 | 7.64 | 1 | 1 | 1 | 1 | 1 |
| k8 | – |  | – | 3 | 9.57 | 1 | 1 | 1 | 1 | 1 |
| L1* | 2-OCH3-phenyl | 4-CH3-phenyl | C=O | – | 7.00 | 1 | 1 | 1 | 1 | 1 |
| L2 | 2-OCH3-phenyl | 4-CH3-phenyl | CHOH | – | 7.40 | 1 | 1 | 1 | 1 | 1 |
| L3 | 2-OCH3-phenyl | 2-CH3-phenyl | C=O | – | 7.60 | 1 | 1 | 1 | 1 | 1 |
| L4* | 2-OCH3-phenyl | 2-CH3-phenyl | CHOH | – | 7.67 | 1 | 1 | 1 | 1 | 1 |
| L5 | 2-OCH3-phenyl | 2,4-di-CH3-phenyl | C=O | – | 7.38 | 1 | 1 | 1 | 1 | 1 |
| L6* | 2-OCH3-phenyl | 2,4-di-CH3-phenyl | CHOH | – | 7.05 | 1 | 1 | 1 | 1 | 1 |
| L7 | 2-OH-phenyl | 2,4-di-CH3-phenyl | C=O | – | 6.94 | 1 | 1 | 1 | 1 | 1 |
| L8 | 2-OH-phenyl | 2,4-di-CH3-phenyl | CHOH | – | 6.96 | 1 | 1 | 1 | 1 | 1 |
| L9 | 1-Naphtyl | 2,4-di-CH3-phenyl | C=O | – | 6.30 | −1 | −1 | −1 | −1 | −1 |
| m1 | – | (CH2)2 | – | – | 8.66 | 1 | 1 | 1 | 1 | 1 |
| m2 | – | (CH2)3 | – | – | 8.25 | 1 | 1 | 1 | 1 | 1 |
| m3 | – | (CH2)4 | – | – | 9.05 | 1 | 1 | 1 | 1 | 1 |
| m4* | – | (CH2)5 | – | – | 8.76 | 1 | 1 | 1 | 1 | 1 |
| m5 | – | (CH2)6 | – | – | 8.80 | 1 | 1 | 1 | 1 | 1 |
| m6 | – | (CH2)8 | – | – | 8.32 | 1 | 1 | 1 | 1 | 1 |
| m7* | – | (CH2)10 | – | – | 7.55 | 1 | 1 | 1 | 1 | 1 |
| m8* | – | (CH2)12 | – | – | 6.60 | −1 | 1** | 1** | 1** | 1** |
| m9* | – |  | – | – | 9.21 | 1 | 1 | 1 | 1 | 1 |
| m10 | – |  | – | – | 8.56 | 1 | 1 | 1 | 1 | 1 |
| n1 | H | 2-CH3OC6H4 | NH | 0 | 7.48 | 1 | 1 | 1 | 1 | 1 |
| n2 | H | 2-CH3OC6H4 | S | 0 | 7.51 | 1 | 1 | 1 | 1 | 1 |
| n3* | H | 2-CH3OC6H4 | O | 0 | 7.17 | 1 | 1 | 1 | 1 | 1 |
| n4* | H | 2-NO2C6H4 | NH | 0 | 7.02 | 1 | 1 | 1 | −1** | −1** |
| n5* | H | 2-NO2C6H4 | S | 0 | 5.77 | −1 | 1** | 1** | −1 | −1 |
| n6* | H | 2-NO2C6H4 | O | 0 | 6.49 | −1 | 1** | 1** | −1 | −1 |
| n7 | H | 2-CH3OC6H4 | NH | 1 | 9.00 | 1 | 1 | 1 | 1 | 1 |
| n8 | H | 2-CH3OC6H4 | S | 1 | 9.54 | 1 | 1 | 1 | 1 | 1 |
| n9 | H | 2-CH3OC6H4 | O | 1 | 9.26 | 1 | 1 | 1 | 1 | 1 |
| n10 | H | 2-NO2C6H4 | NH | 1 | 7.78 | 1 | 1 | 1 | 1 | 1 |
| n11 | H | 2-NO2C6H4 | S | 1 | 8.02 | 1 | 1 | 1 | 1 | 1 |
| n12 | H | 2-NO2C6H4 | O | 1 | 7.85 | 1 | 1 | 1 | 1 | 1 |
| n13 | H | Pyridin-2-yl | S | 1 | 9.11 | 1 | 1 | 1 | 1 | 1 |
| n14* | H | Pyridin-2-yl | O | 1 | 8.80 | 1 | 1 | 1 | 1 | 1 |
| n15 | H | Pyrimidin-2-yl | S | 1 | 8.60 | 1 | 1 | 1 | 1 | 1 |
| n16 | H | Pyrimidin-2-yl | O | 1 | 7.83 | 1 | 1 | 1 | 1 | 1 |
| n17 | H | 2-CH3OC6H4 | NCH3 | 1 | 9.57 | 1 | 1 | 1 | 1 | 1 |
| n18 | Cl | 2-CH3OC6H4 | S | 1 | 9.00 | 1 | 1 | 1 | 1 | 1 |
| n19 | Cl | 2-CH3OC6H4 | O | 1 | 9.06 | 1 | 1 | 1 | 1 | 1 |
| n20* | H | 2-CH3OC6H4 | S | 2 | 9.57 | 1 | 1 | 1 | 1 | 1 |
| n21 | H | 2-CH3OC6H4 | O | 2 | 10.03 | 1 | 1 | 1 | 1 | 1 |
| n22 | H | 2-CH3OC6H4 | S | 4 | 8.89 | 1 | 1 | 1 | 1 | 1 |
| n23 | H | 2-CH3OC6H4 | O | 4 | 9.28 | 1 | 1 | 1 | 1 | 1 |
| Algorithm | Variables | Training set | Test set | ||||
|---|---|---|---|---|---|---|---|
| 1-FNR | 1-FPR | T A | 1-FNR | 1-FPR | TA | ||
| MLR-SVM | MSD,MAXDN,MAXDP,PW4, PW5, PJI2,BAC,ICR | 0.986 | 1.000 | 0.991 | 0.821 | 0.667 | 0.775 |
| MLR-AdaBoost-SVM | 1.000 | 97.5 | 0.991 | 0.893 | 0.667 | 0.825 | |
| PSO-SVM | TI1, D/D, DELS, S1K,ECC, MDDD, BAC | 1.000 | 97.5 | 0.991 | 0.964 | 0.833 | 0.925 |
| PSO-AdaBoost-SVM | 1.000 | 1.000 | 1.000 | 0.964 | 0.917 | 0.950 | |
| Model | Variables | Training set Total | Test set Total | ||||
|---|---|---|---|---|---|---|---|
| %, 1 | %, −1 | Total Accuracy (%) | %, 1 | %, −1 | Total Accuracy (%) | ||
| Constitutional | Se, Mv, ARR, nC | 94.5 | 77.5 | 88.5 | 92.9 | 50.0 | 80.0 |
| RDF | RDF010u, RDF045u, RDF115m, RDF070v, RDF090v, RDF065e, RDF150p | 95.9 | 85.0 | 92.0 | 89.3 | 91.7 | 90.0 |
| Topological | TI1, D/D, DELS, S1K, ECC, MDDD, BAC | 100.0 | 100.0 | 100.0 | 96.4 | 91.7 | 95.0 |
| 2D | ATS8m, ATS1e, MATS4m, GATS4m, GATS1v, GATS4e, GATS5p | 98.6 | 97.5 | 98.2 | 92.9 | 75.0 | 87.5 |
| Geometrical | AGDD, SPAN | 100.0 | 100.0 | 100.0 | 89.3 | 66.7 | 82.5 |
| 3D MoRSE | Mor02u, Mor06u, Mor22m, Mor25m, Mor05v, Mor16v, Mor32v, Mor02e, Mor26p | 98.6 | 95.0 | 97.3 | 96.4 | 66.7 | 87.5 |
| GETAWAY | HTu, H6m, HATS3m, HATS5m, R5u, RTv, R7v+ | 100.0 | 100.0 | 100.0 | 85.7 | 83.3 | 85.0 |
| Information indices | IDET, TIC0, SIC0, TIC5 | 98.6 | 100.0 | 99.1 | 100.0 | 50.0 | 85.0 |
| Randic molecular profiles | DP19, SP01, SP09, SP18 | 97.3 | 75.0 | 89.4 | 96.4 | 66.7 | 87.5 |
| WHIM | L1u, L2v, G3p, Te, Ts | 100.0 | 100.0 | 100.0 | 92.9 | 58.3 | 82.5 |
| Burden eigenvalues | BELm5, BEHv3, BELv1, BELv3, BEHe3 | 89.0 | 72.5 | 83.2 | 89.3 | 66.7 | 82.5 |
| Eigenuaulue based indices | Eig1p, SEigZ, AEige, VEZ2, VRZ1, VRp2 | 100.0 | 100.0 | 100.0 | 92.9 | 75.0 | 87.5 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cheng, Z.; Zhang, Y.; Zhou, C.; Zhang, W.; Gao, S. Classification of 5-HT1A Receptor Ligands on the Basis of Their Binding Affinities by Using PSO-Adaboost-SVM. Int. J. Mol. Sci. 2009, 10, 3316-3337. https://doi.org/10.3390/ijms10083316
Cheng Z, Zhang Y, Zhou C, Zhang W, Gao S. Classification of 5-HT1A Receptor Ligands on the Basis of Their Binding Affinities by Using PSO-Adaboost-SVM. International Journal of Molecular Sciences. 2009; 10(8):3316-3337. https://doi.org/10.3390/ijms10083316
Chicago/Turabian StyleCheng, Zhengjun, Yuntao Zhang, Changhong Zhou, Wenjun Zhang, and Shibo Gao. 2009. "Classification of 5-HT1A Receptor Ligands on the Basis of Their Binding Affinities by Using PSO-Adaboost-SVM" International Journal of Molecular Sciences 10, no. 8: 3316-3337. https://doi.org/10.3390/ijms10083316