Experimental and Computational Characterization of Biological Liquid Crystals: A Review of Single-Molecule Bioassays

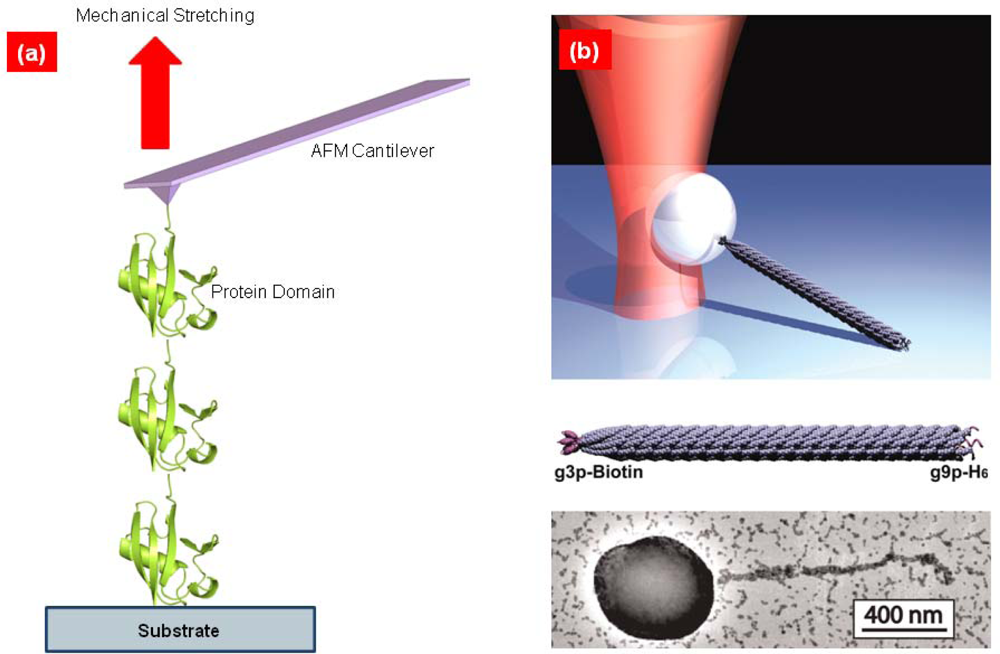{kind=link}
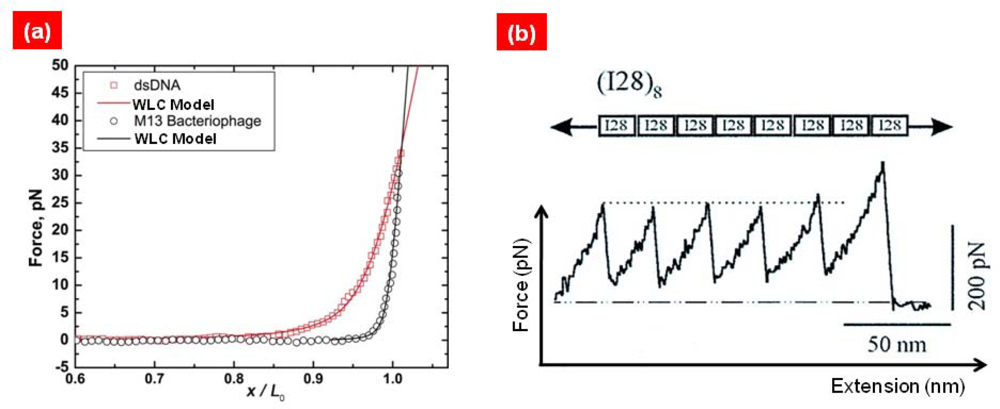{kind=link}
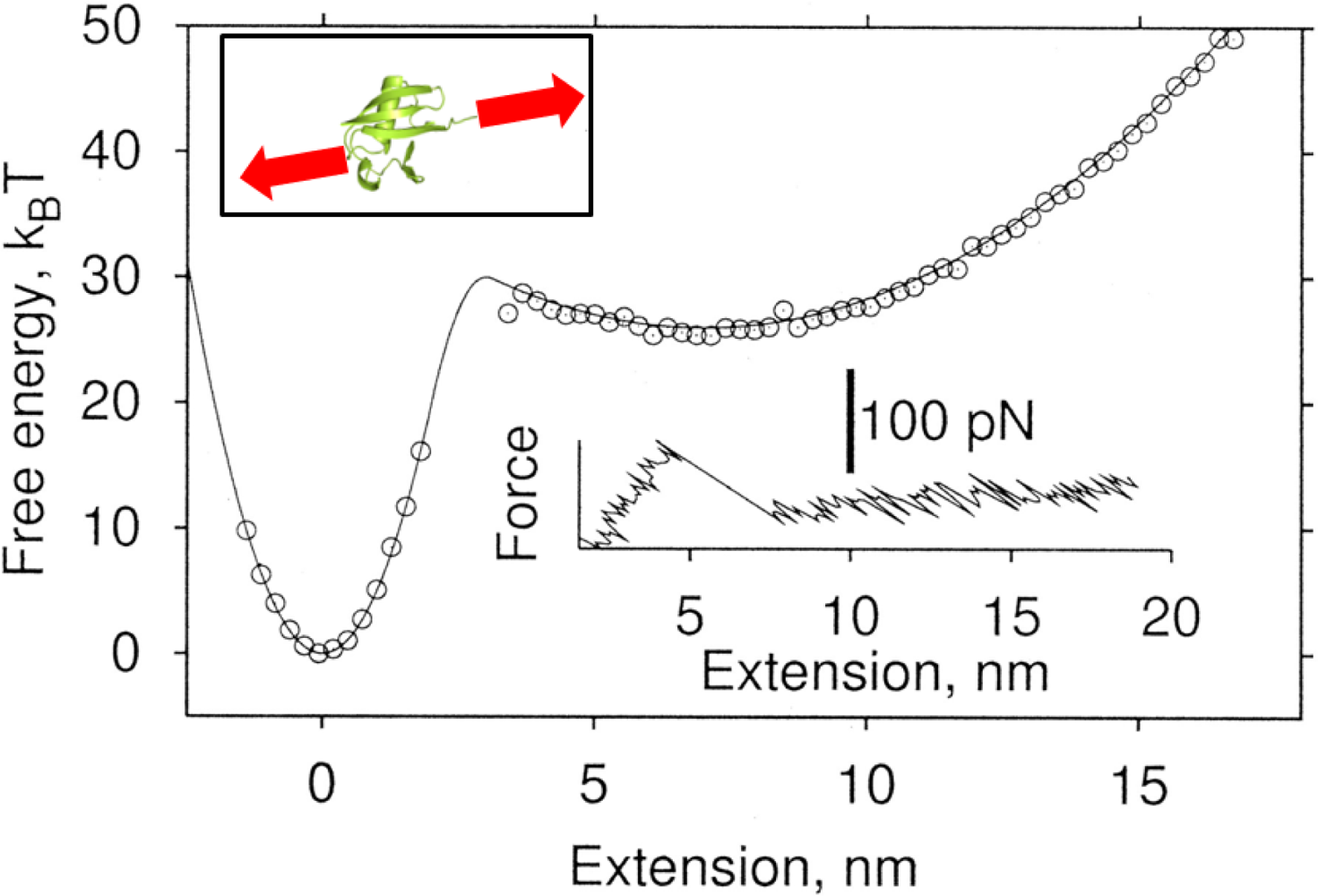{kind=link}
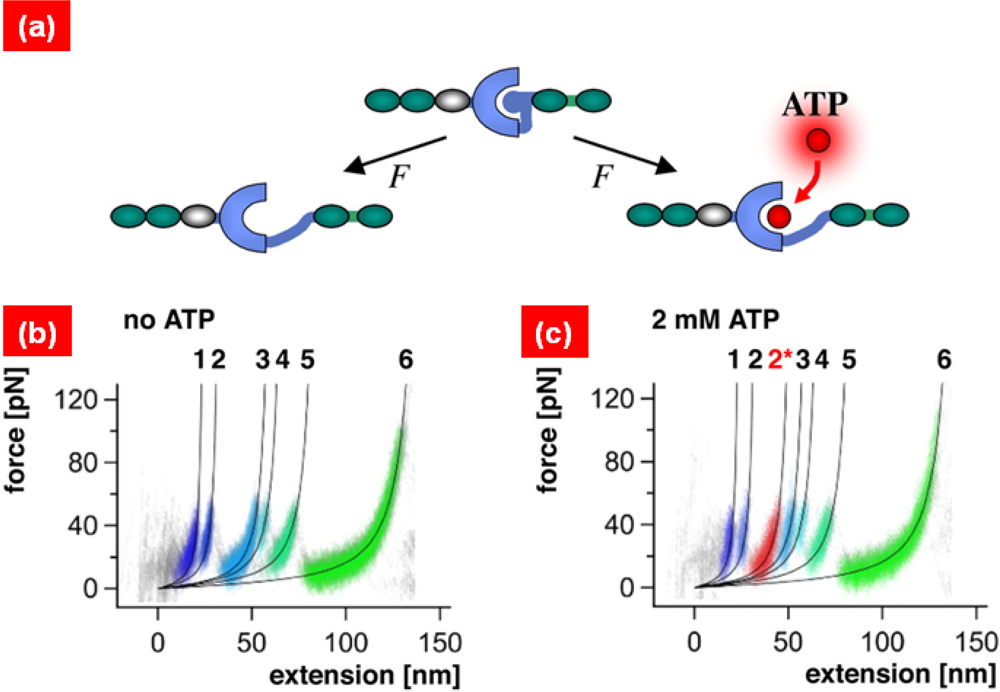{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
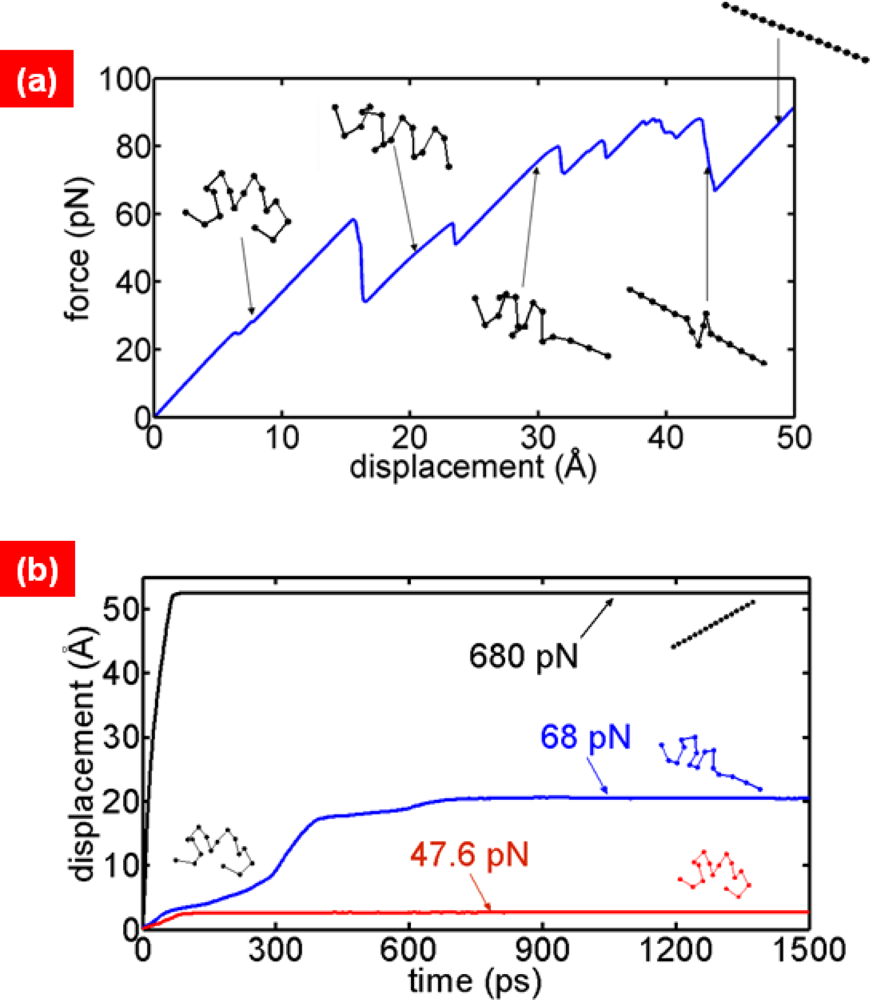
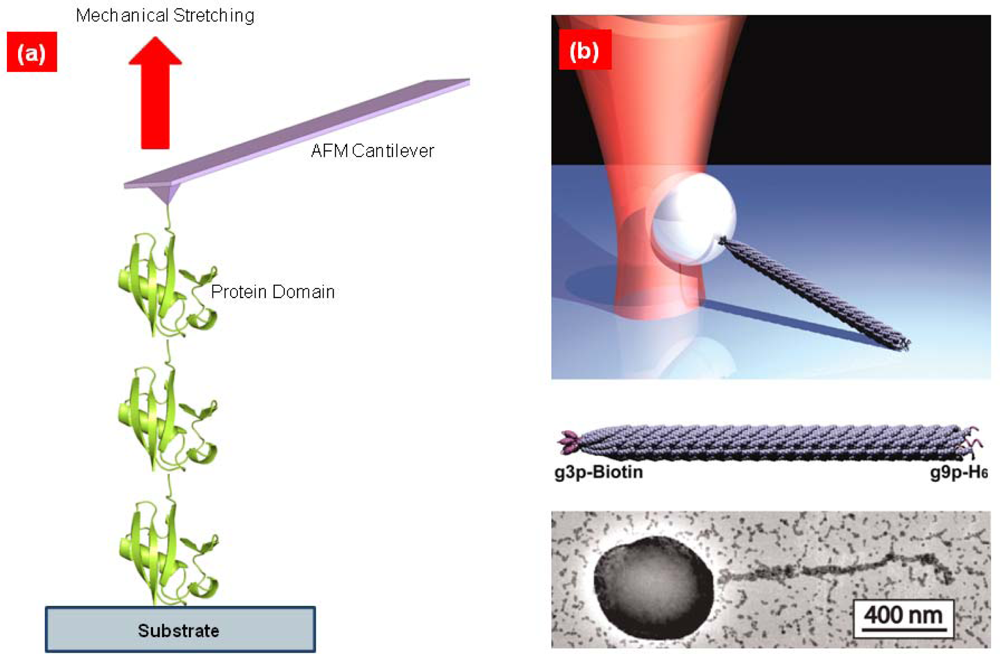
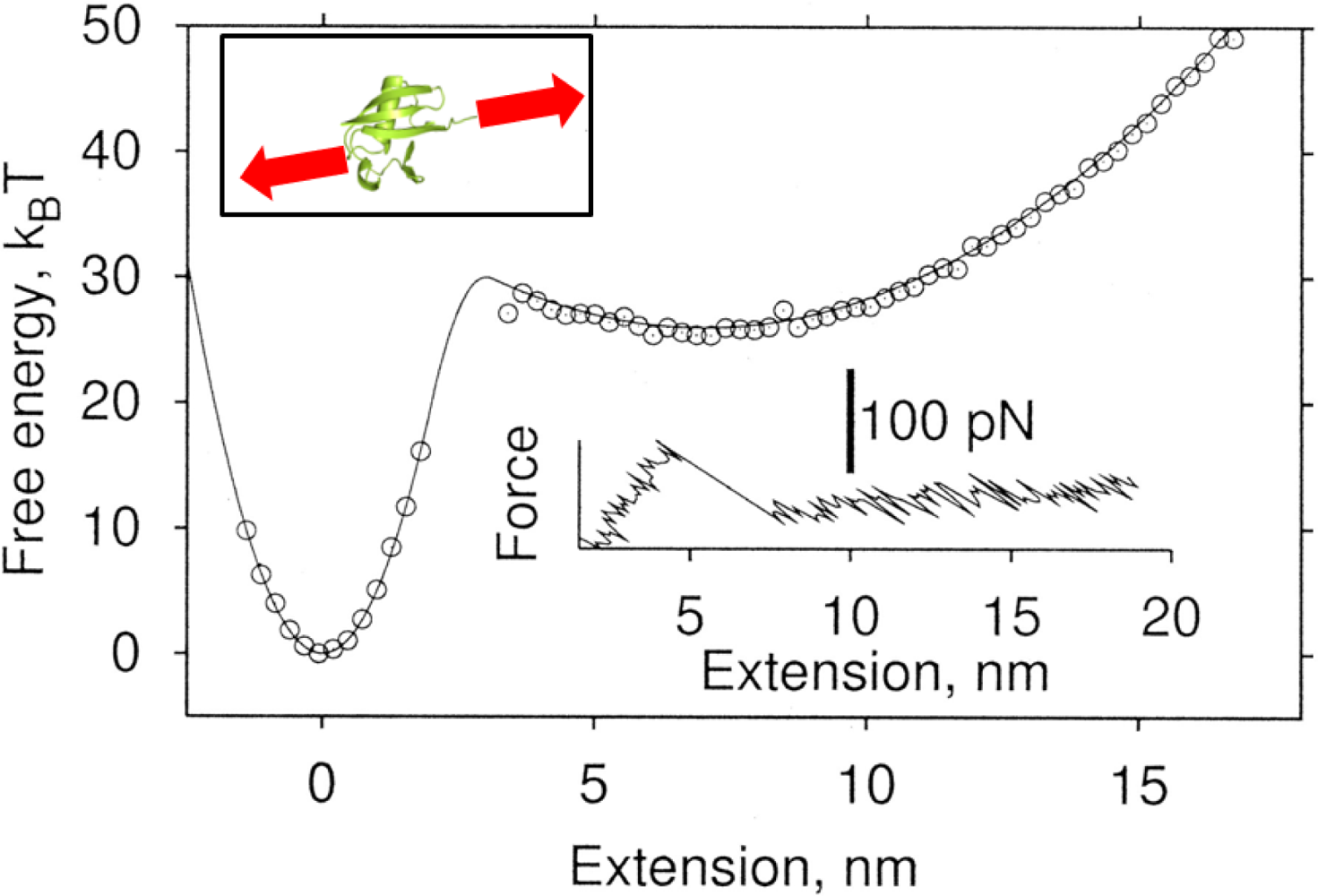
2. Single-Molecule Pulling Experiments
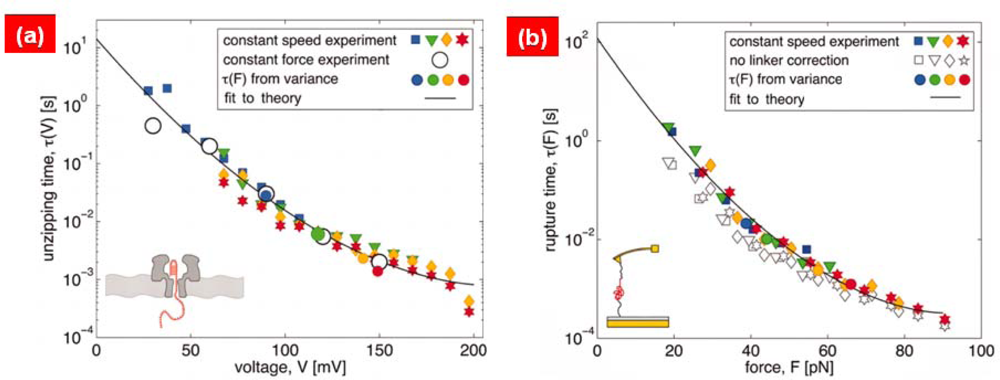
3. Bond Rupture Models: From Bell’s Theory to Dudko-Hummer-Szabo (DHS) Theory
4. Computational Simulations: Coarse-Grained Molecular Dynamics (MD) Simulations
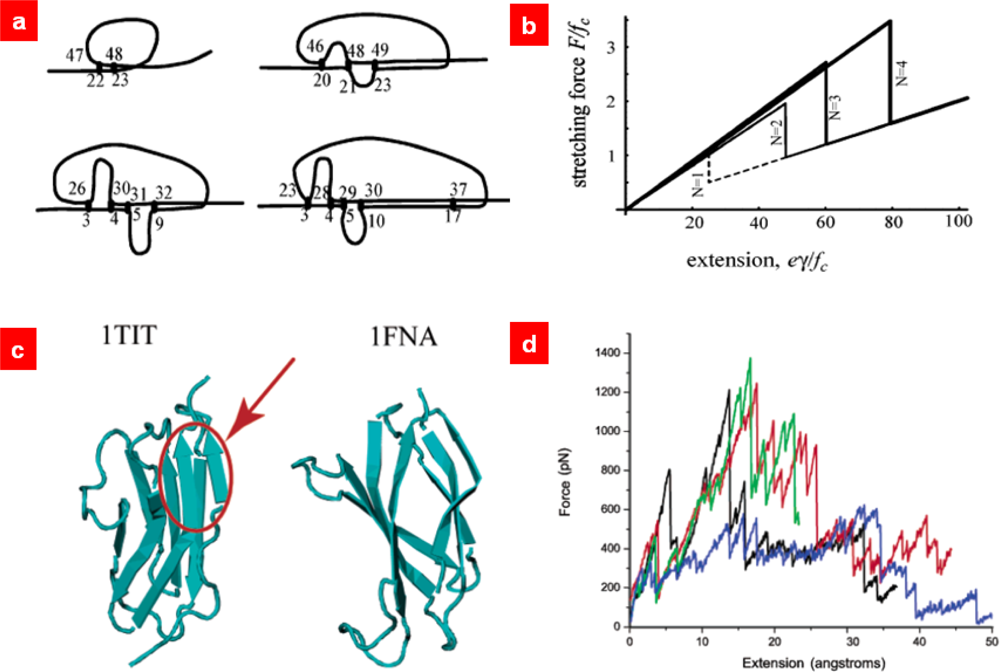
5. Statistical Mechanics-Based Models: Coarse-Grained Chain Molecule Models for Protein Unfolding Mechanics
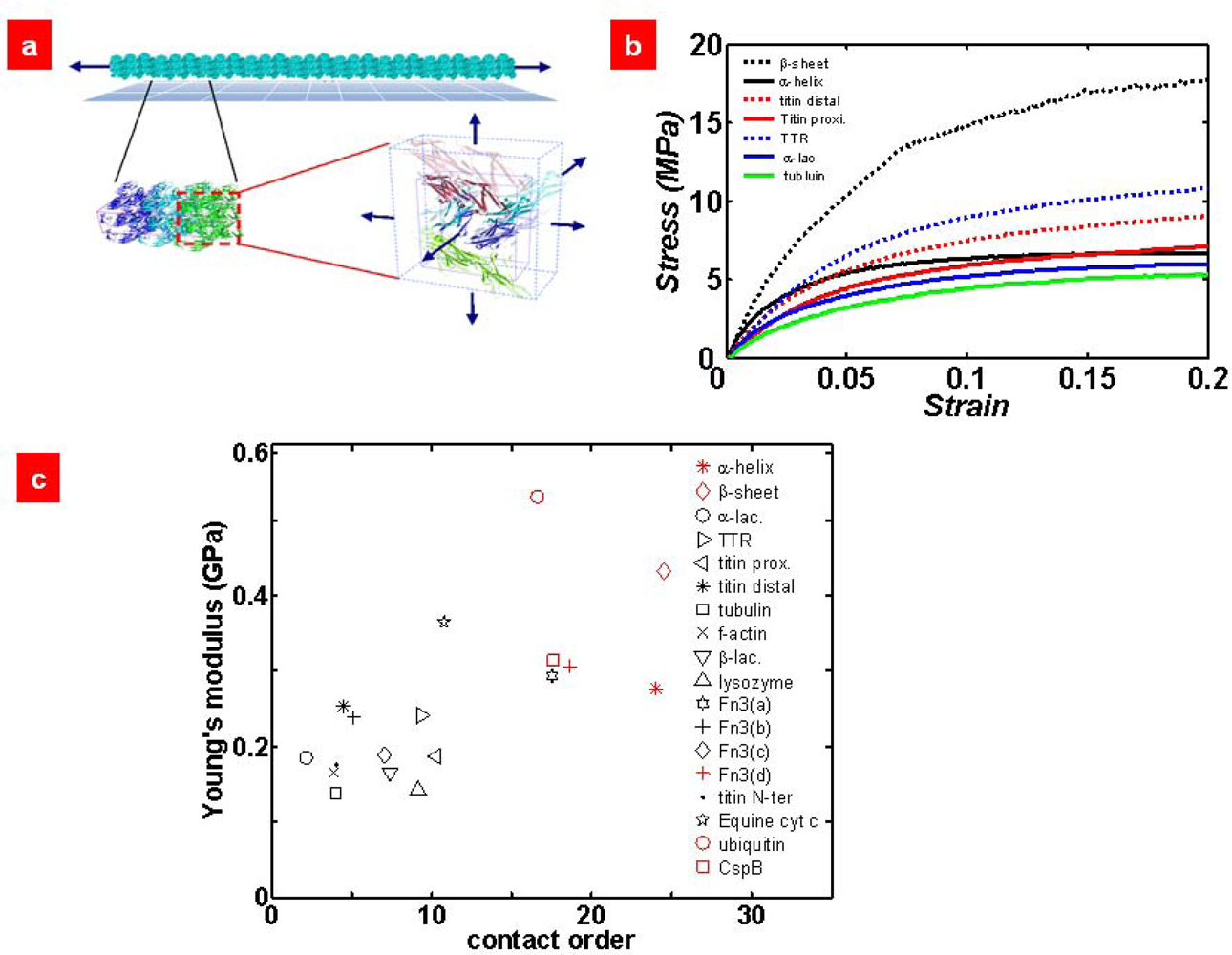
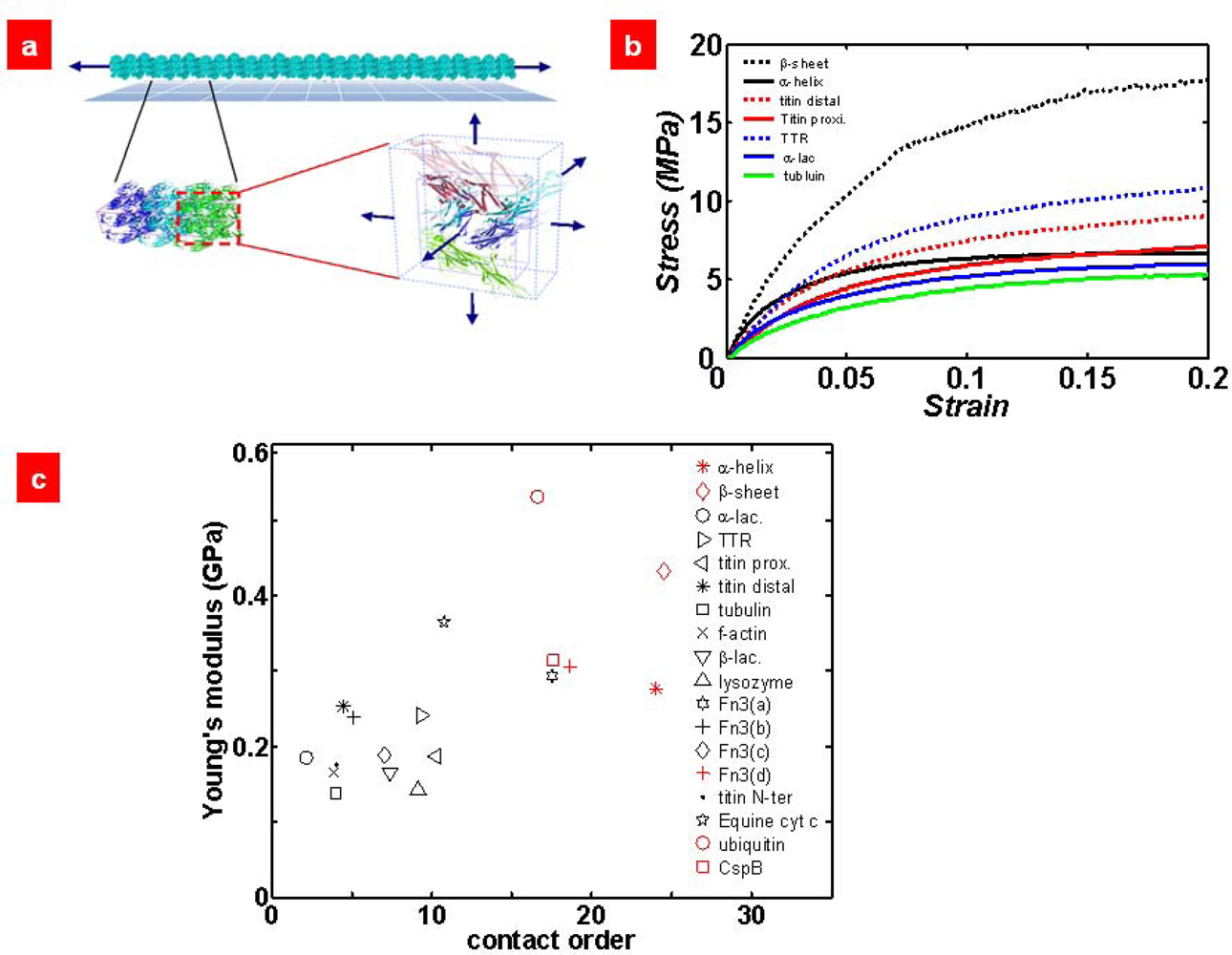
6. Micromechanics Model for Mechanical Characterization
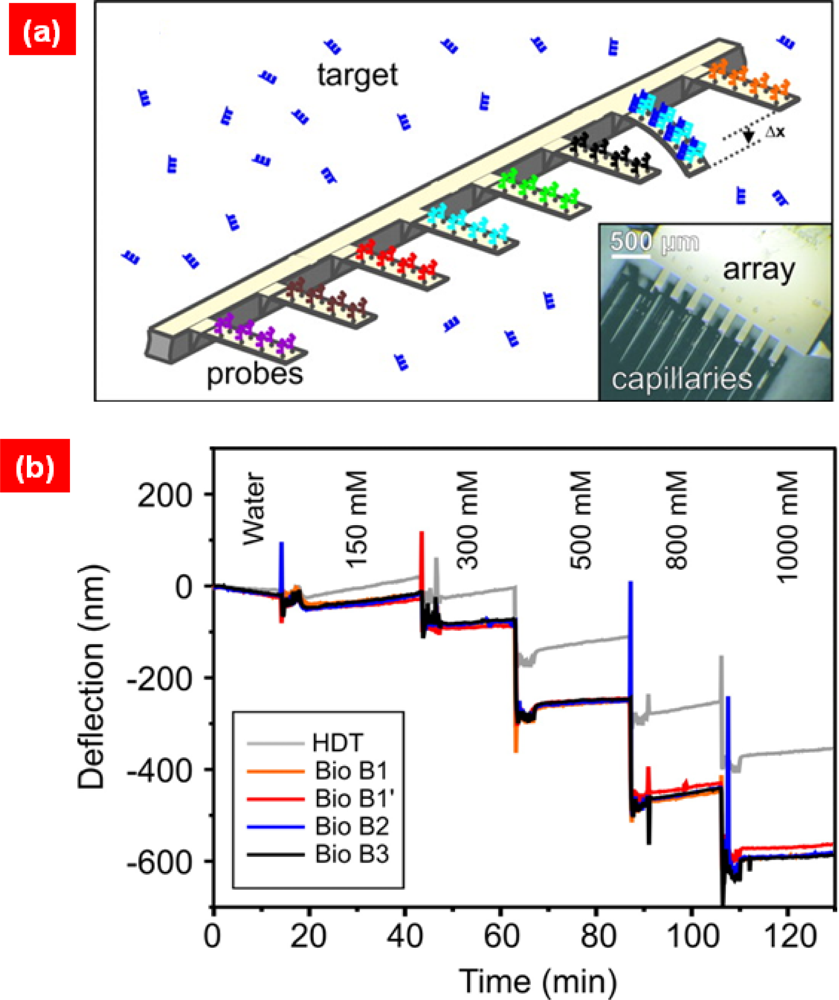
7. Cantilever Bioassay for in Vitro Molecular Recognitions
8. Conclusions
Acknowledgments
References
- Shao, Z; Vollrath, F. Surprising strength of of silkworm silk. Nature 2002, 418, 741. [Google Scholar]
- Porter, D; Vollrath, F. Silk as a biomimetic ideal for structural polymers. Adv. Mater 2009, 21, 487–492. [Google Scholar]
- Borgia, A; Williams, PM; Clarke, J. Single-molecule studies of protein folding. Annu. Rev. Biochem 2008, 77, 101–125. [Google Scholar]
- Neuman, KC; Lionnet, T; Allemand, JF. Single-molecule micromanipulation techniques. Annu. Rev. Mater. Res 2007, 37, 33–67. [Google Scholar]
- Muller, DJ; Dufrene, YF. Atomic force microscopy as a multifunctional molecular toolbox in nanobiotechnology. Nat. Nanotechnol 2008, 3, 261–269. [Google Scholar]
- Bustamante, C; Bryant, Z; Smith, SB. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar]
- Bustamante, C; Chemla, YR; Forde, NR; Izhaky, D. Mechanical processes in biochemistry. Annu. Rev. Biochem 2004, 73, 705–748. [Google Scholar]
- Li, PTX; Vieregg, J; Tinoco, I. How RNA unfolds and refolds. Annu. Rev. Biochem 2008, 77, 77–100. [Google Scholar]
- Joo, C; Balci, H; Ishitsuka, Y; Buranachai, C; Ha, T. Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem 2008, 77, 51–76. [Google Scholar]
- Roy, R; Hohng, S; Ha, T. A practical guide to single-molecule FRET. Nat. Meth 2008, 5, 507–516. [Google Scholar]
- Bustamante, C; Marko, JF; Siggia, ED; Smith, S. Entropic elasticity of lambda-phage DNA. Science 1994, 265, 1599–1600. [Google Scholar]
- Oberhauser, AF; Marszalek, PE; Erickson, HP; Fernandez, JM. The molecular elasticity of the extracellular matrix protein tenascin. Nature 1998, 393, 181–185. [Google Scholar]
- Marszalek, PE; Lu, H; Li, HB; Carrion-Vazquez, M; Oberhauser, AF; Schulten, K; Fernandez, JM. Mechanical unfolding intermediates in titin modules. Nature 1999, 402, 100–103. [Google Scholar]
- Evans, E. Probing the relation between force-life time and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct 2001, 30, 105–128. [Google Scholar]
- Evans, EA; Calderwood, DA. Forces and bond dynamics in cell adhesion. Science 2007, 316, 1148–1153. [Google Scholar]
- Lu, H; Isralewitz, B; Krammer, A; Vogel, V; Schulten, K. Unfolding of titin immunoglobulin domains by steered molecular dynamics simulation. Biophys. J 1998, 75, 662–671. [Google Scholar]
- Lu, H; Schulten, K. Steered molecular dynamics simulations of force-induced protein domain unfolding. Proteins 1999, 35, 453–463. [Google Scholar]
- Gao, M; Craig, D; Vogel, V; Schulten, K. Identifying unfolding intermediates of FN-III(10) by steered molecular dynamics. J. Mol. Biol 2002, 323, 939–950. [Google Scholar]
- Sotomayor, M; Schulten, K. Single-molecule experiments in vitro and in silico. Science 2007, 316, 1144–1148. [Google Scholar]
- Elber, R. Long-timescale simulation methods. Curr. Opin. Struct. Biol 2005, 15, 151–156. [Google Scholar]
- Cieplak, M; Hoang, TX; Robbins, MO. Folding and stretching in a Go-like model of titin. Proteins: Struct. Funct. Genet 2002, 49, 114–124. [Google Scholar]
- Cieplak, M; Hoang, TX; Robbins, MO. Thermal folding and mechanical unfolding pathways of protein secondary structures. Proteins: Struct. Funct. Genet 2002, 49, 104–113. [Google Scholar]
- Cieplak, M; Hoang, TX; Robbins, MO. Thermal effects in stretching of Go-like models of titin and secondary structures. Proteins: Struct. Funct. Bioinfo 2004, 56, 285–297. [Google Scholar]
- Eom, K; Yoon, G; Kim, J-I; Na, S. Coarse-grained elastic models of protein structures for understanding their mechanics and/or dynamics. J Comput Theor Nanosci. in press.
- Eom, K; Na, S. Coarse-grained structural model of protein molecules. In Computational Biology: New Research; Russe, AS, Ed.; Nova Science Publisher: New York, NY, USA, 2009; pp. 193–213. [Google Scholar]
- Kassner, ME; Nemat-Nasser, S; Suo, ZG; Bao, G; Barbour, JC; Brinson, LC; Espinosa, H; Gao, HJ; Granick, S; Gumbsch, P; Kim, KS; Knauss, W; Kubin, L; Langer, J; Larson, BC; Mahadevan, L; Majumdar, A; Torquato, S; van Swol, F. New directions in mechanics. Mech. Mater 2005, 37, 231–259. [Google Scholar]
- Waggoner, PS; Craighead, HG. Micro- and nanomechanical sensors for environmental, chemical, and biological detection. Lab Chip 2007, 7, 1238–1255. [Google Scholar]
- Berger, R; Delamarche, E; Lang, HP; Gerber, C; Gimzewski, JK; Meyer, E; Guntherodt, HJ. Surface stress in the self-assembly of alkanethiols on gold. Science 1997, 276, 2021–2024. [Google Scholar]
- Khalil, AS; Ferrer, JM; Brau, RR; Kottmann, ST; Noren, CJ; Lang, MJ; Belcher, AM. From the cover: Single M13 bacteriophage tethering and stretching. Proc. Natl. Acad. Sci. USA 2007, 104, 4892–4897. [Google Scholar]
- Yamakawa, H; Fujii, M. Wormlike chains near the rod limit: Path integral in the WKB approximation. J. Chem. Phys 1973, 59, 6641–6644. [Google Scholar]
- Strick, T; Allemand, JF; Croquette, V; Bensimon, D. Twisting and stretching single DNA molecules. Prog. Biophys. Mol. Biol 2000, 74, 115–140. [Google Scholar]
- Smith, DE; Tans, SJ; Smith, SB; Grimes, S; Anderson, DE; Bustamante, C. The bacteriophage 29 portal motor can package DNA against a large internal force. Nature 2001, 413, 748–752. [Google Scholar]
- Gore, J; Bryant, Z; Nollmann, M; Le, MU; Cozzarelli, NR; Bustamante, C. DNA overwinds when stretched. Nature 2006, 442, 836–839. [Google Scholar]
- Seol, Y; Li, JY; Nelson, PC; Perkins, TT; Betterton, MD. Elasticity of short DNA molecules: Theory and experiment for contour lengths of 0.6–7 mu m. Biophys. J 2007, 93, 4360–4373. [Google Scholar]
- Liphardt, J; Onoa, B; Smith, SB; Tinoco, I; Bustamante, C. Reversible unfolding of single RNA molecules by mechanical force. Science 2001, 292, 733–737. [Google Scholar]
- Cheng, W; Dumont, S; Tinoco, I, Jr; Bustamante, C. From the cover: NS3 helicase actively separates RNA strands and senses sequence barriers ahead of the opening fork. Proc. Natl. Acad. Sci. USA 2007, 104, 13954–13959. [Google Scholar]
- Li, PTX; Bustamante, C; Tinoco, I, Jr. Real-time control of the energy landscape by force directs the folding of RNA molecules. Proc. Natl. Acad. Sci. USA 2007, 104, 7039–7044. [Google Scholar]
- Ke, C; Humeniuk, M; S-Gracz, H; Marszalek, PE. Direct measurements of base stacking interactions in DNA by single-molecule atomic-force spectroscopy. Phys. Rev. Lett 2007, 99, 018302. [Google Scholar]
- Ke, CH; Loksztejn, A; Jiang, Y; Kim, M; Humeniuk, M; Rabbi, M; Marszalek, PE. Detecting solvent-driven transitions of poly(A) to double-stranded conformations by atomic force microscopy. Biophys. J 2009, 96, 2918–2925. [Google Scholar]
- Rief, M; Fernandez, JM; Gaub, HE. Elastically coupled two-level systems as a model for biopolymer extensibility. Phys. Rev. Lett 1998, 81, 4764–4767. [Google Scholar]
- Oberhauser, AF; Marszalek, PE; Carrion-Vazquez, M; Fernandez, JM. Single protein misfolding events captured by atomic force microscopy. Nat. Struct. Mol. Biol 1999, 6, 1025–1028. [Google Scholar]
- Klimov, DK; Thirumalai, D. Native topology determines force-induced unfolding pathways in globular proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 7254–7259. [Google Scholar]
- Dietz, H; Rief, M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc. Natl. Acad. Sci. USA 2004, 101, 16192–16197. [Google Scholar]
- Dietz, H; Berkemeier, F; Bertz, M; Rief, M. Anisotropic deformation response of single protein molecules. Proc. Natl. Acad. Sci. USA 2006, 103, 12724–12728. [Google Scholar]
- Mickler, M; Dima, RI; Dietz, H; Hyeon, C; Thirumalai, D; Rief, M. Revealing the bifurcation in the unfolding pathways of GFP by using single-molecule experiments and simulations. Proc. Natl. Acad. Sci. USA 2007, 104, 20268–20273. [Google Scholar]
- Bertz, M; Wilmanns, M; Rief, M. The titin-telethonin complex is a directed, superstable molecular bond in the muscle Z-disk. Proc. Natl. Acad. Sci. USA 2009, 106, 13307–133310. [Google Scholar]
- Ke, CH; Jiang, Y; Rivera, M; Clark, RL; Marszalek, PE. Pulling geometry-induced errors in single molecule force spectroscopy measurements. Biophys. J 2007, 92, L76–L78. [Google Scholar]
- Rivera, M; Lee, W; Ke, C; Marszalek, PE; Cole, DG; Clark, RL. Minimizing pulling geometry errors in atomic force microscope single molecule force spectroscopy. Biophys. J 2008, 95, 3991–3998. [Google Scholar]
- Rajesh, R; Giri, D; Jensen, I; Kumar, S. Role of pulling direction in understanding the energy landscape of proteins. Phys. Rev. E 2008, 78, 021905. [Google Scholar]
- Liphardt, J; Dumont, S; Smith, SB; Tinoco, I; Bustamante, C. Equilibrium information from nonequilibrium measurements in an experimental test of Jarzynski's equality. Science 2002, 296, 1832–1835. [Google Scholar]
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett 1997, 78, 2690–2693. [Google Scholar]
- Hummer, G; Szabo, A. Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl. Acad. Sci. USA 2001, 98, 3658–3661. [Google Scholar]
- Harris, NC; Song, Y; Kiang, C-H. Experimental free energy surface reconstruction from single-molecule force spectroscopy using Jarzynski's equality. Phys. Rev. Lett 2007, 99, 068101. [Google Scholar]
- Collin, D; Ritort, F; Jarzynski, C; Smith, SB; Tinoco, I; Bustamante, C. Verification of the Crooks fluctuation theorem and recovery of RNA folding free energies. Nature 2005, 437, 231–234. [Google Scholar]
- del Rio, A; Perez-Jimenez, R; Liu, R; Roca-Cusachs, P; Fernandez, JM; Sheetz, MP. Stretching single talin rod molecules activates vinculin binding. Science 2009, 323, 638–641. [Google Scholar]
- Puchner, EM; Alexandrovich, A; Kho, AL; Hensen, U; Schaffer, LV; Brandmeier, B; Grater, F; Grubmuller, H; Gaub, HE; Gautel, M. Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. USA 2008, 105, 13385–13390. [Google Scholar]
- Bell, GI. Models for the specific adhesion of cells to cell. Science 1978, 200, 618–627. [Google Scholar]
- Frauenfelder, H; Sligar, SG; Wolynes, PG. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar]
- Evans, E; Ritchie, K. Dynamic strength of molecular adhesion bonds. Biophys. J 1997, 72, 1541–1555. [Google Scholar]
- Kramers, HA. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 1940, 7, 284–304. [Google Scholar]
- Doi, M; Edwards, SF. The Theory of Polymer Dynamics; Oxford University Press: New York, NY, USA, 1986. [Google Scholar]
- Hyeon, C; Thirumalai, D. Measuring the energy landscape roughness and the transition state location of biomolecules using single molecule mechanical unfolding experiments. J. Phys.: Condens. Matter 2007, 19, 113101. [Google Scholar]
- Freund, LB. Characterizing the resistance generated by a molecular bond as it is forcibly separated. Proc. Natl. Acad. Sci. USA 2009, 106, 8818–8823. [Google Scholar]
- Dudko, OK; Filippov, AE; Klafter, J; Urbakh, M. Beyond the conventional description of dynamic force spectroscopy of adhesion bonds. Proc. Natl. Acad. Sci. USA 2003, 100, 11378–11381. [Google Scholar]
- Garg, A. Escape-field distribution for escape from a metastable potential well subject to a steadily increasing bias field. Phys. Rev. B 1995, 51, 15592–15595. [Google Scholar]
- Dudko, OK; Hummer, G; Szabo, A. Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys. Rev. Lett 2006, 96, 108101. [Google Scholar]
- Dudko, OK; Hummer, G; Szabo, A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl. Acad. Sci. USA 2008, 105, 15755–15760. [Google Scholar]
- McCammon, JA; Harvey, S. Dynamics of Proteins and Nucleic Acids; Cambridge University Press: Cambridge, UK, 1987. [Google Scholar]
- Karplus, M; Petsko, GA. Molecular dynamics simulations in biology. Nature 1990, 347, 631–639. [Google Scholar]
- Karplus, M; McCammon, JA. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol 2002, 9, 646–652. [Google Scholar]
- Phillips, R; Dittrich, M; Schulten, K. Quasicontinuum representation of atomic-scale mechanics: From proteins to dislocations. Annu. Rev. Mater. Res 2002, 32, 219–233. [Google Scholar]
- Sulkowska, JI; Sulkowski, P; Szymczak, P; Cieplak, M. Stabilizing effect of knots on proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 19714–19719. [Google Scholar]
- Sulkowska, JI; Kloczkowski, A; Sen, TZ; Cieplak, M; Jernigan, RL. Predicting the order in which contacts are broken during single molecule protein stretching experiments. Proteins: Struct. Funct. Bioinfo 2008, 71, 45–60. [Google Scholar]
- Ueda, Y; Taketomi, H; Go, N. Studies on protein folding, unfolding, and fluctuations by computer simulation. II. A. Three-dimensional lattice model of lysozyme. Biopolymers 1978, 17, 1531–1548. [Google Scholar]
- Ueda, Y; Go, N. Theory of large-amplitude conformational fluctuations in native globular proteins-independent fluctuating site model. Int. J. Pept. Protein Res 1976, 8, 551–558. [Google Scholar]
- Go, M; Go, N. Fluctuations of an alpha-helix. Biopolymers 1976, 15, 1119–1127. [Google Scholar]
- Noguti, T; Go, N. Collective variable descrption of small-amplitude conformational fluctuations in a globular protein. Nature 1982, 296, 776–778. [Google Scholar]
- Hayward, S; Go, N. Collective variable description of native protein dynamics. Annu. Rev. Phys. Chem 1995, 46, 223–250. [Google Scholar]
- Hyeon, C; Thirumalai, D. Can energy landscape roughness of proteins and RNA be measured by using mechanical unfolding experiments? Proc. Natl. Acad. Sci. USA 2003, 100, 10249–10253. [Google Scholar]
- Hyeon, C; Thirumalai, D. Mechanical unfolding of RNA: From hairpins to structures with internal multiloops. Biophys. J 2007, 92, 731–743. [Google Scholar]
- Dima, RI; Joshi, H. Probing the origin of tubulin rigidity with molecular simulations. Proc. Natl. Acad. Sci. USA 2008, 105, 15743–15748. [Google Scholar]
- Ermak, DL; McCammon, JA. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys 1978, 69, 1352–1360. [Google Scholar]
- Li, MS; Gabovich, AM; Voitenko, AI. New method for deciphering free energy landscape of three-state proteins. J. Chem. Phys 2008, 129, 105102. [Google Scholar]
- Yew, ZT; McLeish, T; Paci, E. New dynamical window onto the landscape for forced protein unfolding. Phys Rev Lett 2008, 101, 248104.1–248104.4. [Google Scholar]
- Eom, K; Li, PC; Makarov, DE; Rodin, GJ. Relationship between the mechanical properties and topology of cross-linked polymer molecules: Parallel strands maximize the strength of model polymers and protein domains. J. Phys. Chem. B 2003, 107, 8730–8733. [Google Scholar]
- Eom, K; Makarov, DE; Rodin, GJ. Theoretical studies of the kinetics of mechanical unfolding of cross-linked polymer chains and their implications for single-molecule pulling experiments. Phys. Rev. E 2005, 71, 021904. [Google Scholar]
- Kreuzer, HJ; Payne, SH; Livadaru, L. Stretching a macromolecule in an atomic force microscope: Statistical mechanical analysis. Biophys. J 2001, 80, 2505–2514. [Google Scholar]
- Hanke, F; Kreuzer, HJ. Nonequilibrium theory of polymer stretching based on the master equation. Phys. Rev. E 2005, 72, 031805. [Google Scholar]
- Staple, DB; Payne, SH; Reddin, ALC; Kreuzer, HJ. Model for stretching and unfolding the giant multidomain muscle protein using single-molecule force spectroscopy. Phys. Rev. Lett 2008, 101, 248301. [Google Scholar]
- Makarov, DE. A theoretical model for the mechanical unfolding of repeat proteins. Biophys. J 2009, 96, 2160–2167. [Google Scholar]
- Buehler, MJ. Mechanics of protein crystals: Atomistic modeling of elasticity and fracture. J. Comput. Theor. Nanosci 2006, 3, 670–683. [Google Scholar]
- Buehler, MJ. Large-scale hierarhical molecular modeling of nanostructured biological materials. J. Comput. Theor. Nanosci 2006, 3, 603–623. [Google Scholar]
- Yoon, G; Park, H-J; Na, S; Eom, K. Mesoscopic model for mechanical characterization of biological protein materials. J. Comput. Chem 2009, 30, 873–880. [Google Scholar]
- Zhou, M. A new look at the atomistic virial stress: On continuum-molecular system equivalence. Proc. R. Soc. Lond. A 2003, 459, 2347–2392. [Google Scholar]
- Stoney, GG. The tension of metallic films deposited by electrolysis. Proc. R. Soc. Lond. A 1909, 82, 172–175. [Google Scholar]
- Fritz, J; Baller, MK; Lang, HP; Rothuizen, H; Vettiger, P; Meyer, E; Guntherodt, HJ; Gerber, C; Gimzewski, JK. Translating biomolecular recognition into nanomechanics. Science 2000, 288, 316–318. [Google Scholar]
- McKendry, R; Zhang, JY; Arntz, Y; Strunz, T; Hegner, M; Lang, HP; Baller, MK; Certa, U; Meyer, E; Guntherodt, HJ; Gerber, C. Multiple label-free biodetection and quantitative DNA-binding assays on a nanomechanical cantilever array. Proc. Natl. Acad. Sci. USA 2002, 99, 9783–9788. [Google Scholar]
- Wu, GH; Ji, HF; Hansen, K; Thundat, T; Datar, R; Cote, R; Hagan, MF; Chakraborty, AK; Majumdar, A. Origin of nanomechanical cantilever motion generated from biomolecular interactions. Proc. Natl. Acad. Sci. USA 2001, 98, 1560–1564. [Google Scholar]
- Wu, GH; Datar, RH; Hansen, KM; Thundat, T; Cote, RJ; Majumdar, A. Bioassay of prostate-specific antigen (PSA) using microcantilevers. Nat. Biotechnol 2001, 19, 856–860. [Google Scholar]
- Ndieyira, JW; Watari, M; Barrera, AD; Zhou, D; Vogtli, M; Batchelor, M; Cooper, MA; Strunz, T; Horton, MA; Abell, C; Rayment, T; Aeppli, G; McKendry, RA. Nanomechanical detection of antibiotic-mucopeptide binding in a model for superbug drug resistance. Nat. Nanotechnol 2008, 3, 691–696. [Google Scholar]
- Braun, T; Ghatkesar, MK; Backmann, N; Grange, W; Boulanger, P; Letellier, L; Lang, H-P; Bietsch, A; Gerber, C; Hegner, M. Quantitative time-resolved measurement of membrane protein-ligand interactions using microcantilever array sensors. Nat. Nanotechnol 2009, 4, 179–185. [Google Scholar]
- Ghatkesar, MK; Lang, HP; Gerber, C; Hegner, M; Braun, T. Comprehensive characterization of molecular interactions based on nanomechanics. PLoS ONE 2008, 3, e3610. [Google Scholar]
- Braun, T; Barwich, V; Ghatkesar, MK; Bredekamp, AH; Gerber, C; Hegner, M; Lang, HP. Micromechanical mass sensors for biomolecular detection in a physiological environment. Phys. Rev. E 2005, 72, 031907. [Google Scholar]
- Eom, K; Kwon, TY; Yoon, DS; Lee, HL; Kim, TS. Dynamical response of nanomechanical resonators to biomolecular interactions. Phys. Rev. B 2007, 76, 113408. [Google Scholar]
- Yang, YT; Callegari, C; Feng, XL; Ekinci, KL; Roukes, ML. Zeptogram-scale nanomechanical mass sensing. Nano Lett 2006, 6, 583–586. [Google Scholar]
- Naik, AK; Hanay, MS; Hiebert, WK; Feng, XL; Roukes, ML. Towards single-molecule nanomechanical mass spectrometry. Nat. Nanotechnol 2009, 4, 445–450. [Google Scholar]
- Verbridge, SS; Bellan, LM; Parpia, JM; Craighead, HG. Optically driven resonance of nanoscale flexural oscillators in liquid. Nano Lett 2006, 6, 2109–2114. [Google Scholar]
- Kwon, TY; Eom, K; Park, JH; Yoon, DS; Kim, TS; Lee, HL. In situ real-time monitoring of biomolecular interactions based on resonating microcantilevers immersed in a viscous fluid. Appl. Phys. Lett 2007, 90, 223903. [Google Scholar]
- Kwon, T; Eom, K; Park, J; Yoon, DS; Lee, HL; Kim, TS. Micromechanical observation of the kinetics of biomolecular interactions. Appl. Phys. Lett 2008, 93, 173901. [Google Scholar]
- Kwon, T; Park, J; Yang, J; Yoon, DS; Na, S; Kim, C-W; Suh, JS; Huh, YM; Haam, S; Eom, K. Nanomechanical in situ monitoring of proteolysis by cathepsin B. PLoS ONE 2009, 4, e6248. [Google Scholar]
- Burg, TP; Manalis, SR. Suspended microchannel resonators for biomolecular detection. Appl. Phys. Lett 2003, 83, 2698–2700. [Google Scholar]
- Burg, TP; Godin, M; Knudsen, SM; Shen, W; Carlson, G; Foster, JS; Babcock, K; Manalis, SR. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature 2007, 446, 1066–1069. [Google Scholar]
- Burg, TP; Sader, JE; Manalis, SR. Nonmonotonic energy dissipation in microfluidic resonators. Phys. Rev. Lett 2009, 102, 228103. [Google Scholar]







© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Eom, K.; Yang, J.; Park, J.; Yoon, G.; Sohn, Y.S.; Park, S.; Yoon, D.S.; Na, S.; Kwon, T. Experimental and Computational Characterization of Biological Liquid Crystals: A Review of Single-Molecule Bioassays. Int. J. Mol. Sci. 2009, 10, 4009-4032. https://doi.org/10.3390/ijms10094009
Eom K, Yang J, Park J, Yoon G, Sohn YS, Park S, Yoon DS, Na S, Kwon T. Experimental and Computational Characterization of Biological Liquid Crystals: A Review of Single-Molecule Bioassays. International Journal of Molecular Sciences. 2009; 10(9):4009-4032. https://doi.org/10.3390/ijms10094009
Chicago/Turabian StyleEom, Kilho, Jaemoon Yang, Jinsung Park, Gwonchan Yoon, Young Soo Sohn, Shinsuk Park, Dae Sung Yoon, Sungsoo Na, and Taeyun Kwon. 2009. "Experimental and Computational Characterization of Biological Liquid Crystals: A Review of Single-Molecule Bioassays" International Journal of Molecular Sciences 10, no. 9: 4009-4032. https://doi.org/10.3390/ijms10094009