The Diverse Applications of Cladistic Analysis of Molecular Evolution, with Special Reference to Nested Clade Analysis

Abstract
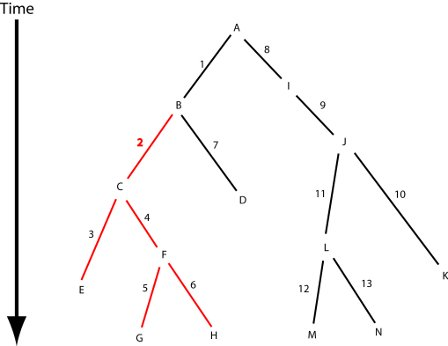
:
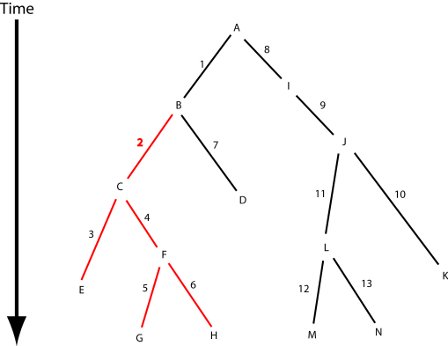{kind=link}
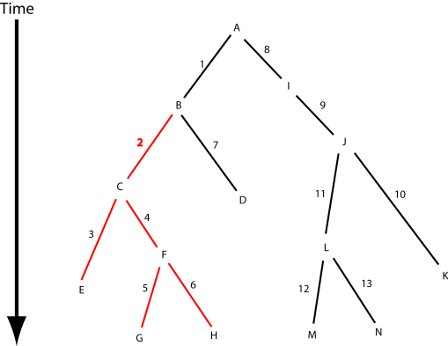{kind=link}
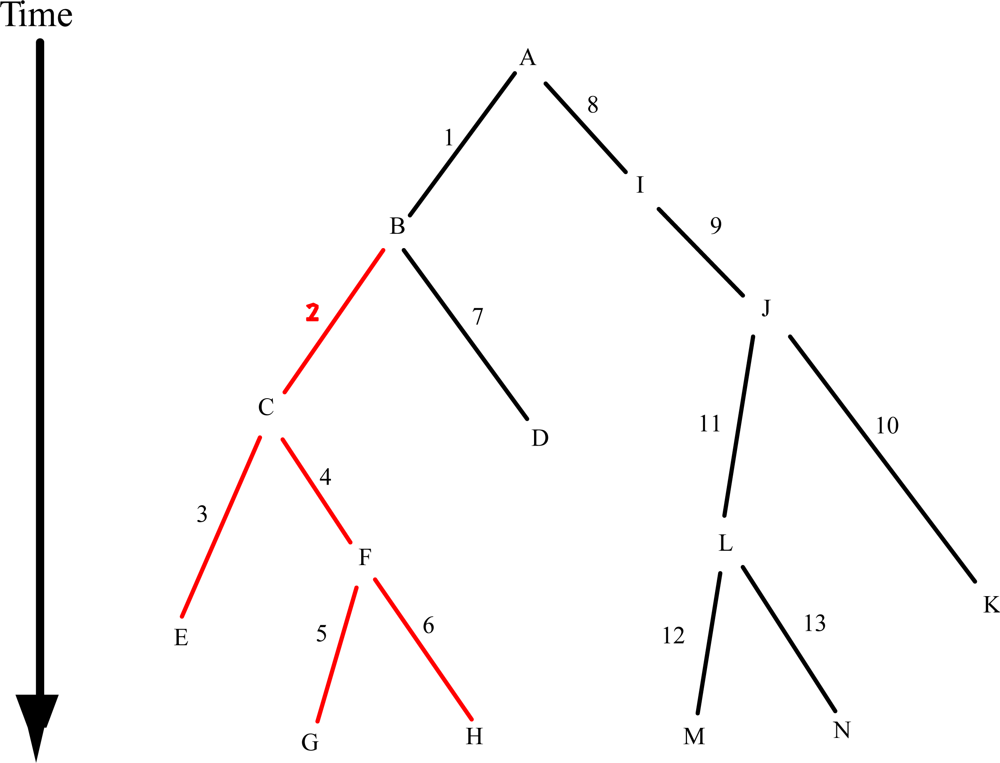{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Genotype/Phenotype Associations
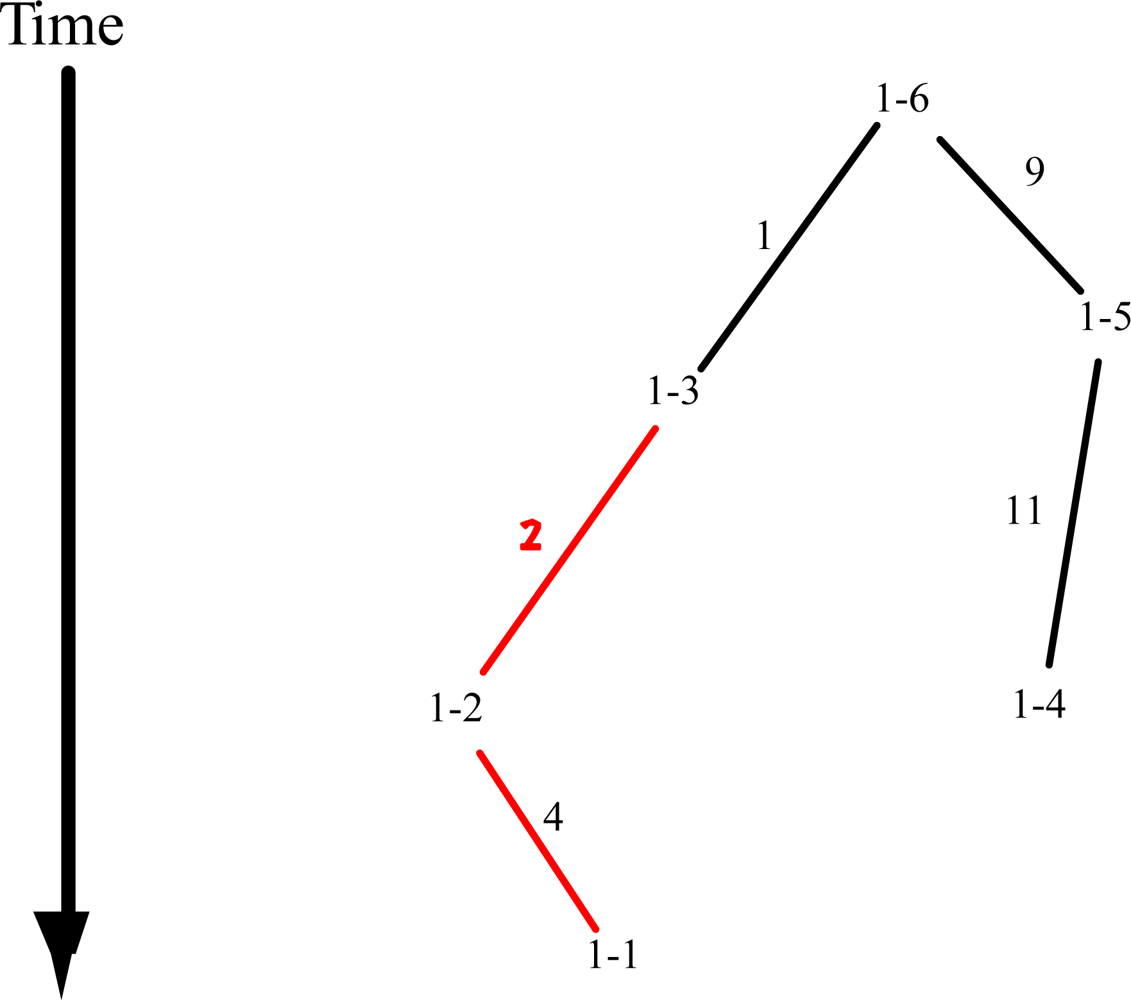
2.1. Basic Rationale for a Cladistic Approach
2.2. Nested Clade Analysis
2.3. Other Evolutionary Methods for Genotype/Phenotype Associations
3. Phylogeography and Associated Applications
3.1. Nested Clade Phylogeographic Analysis
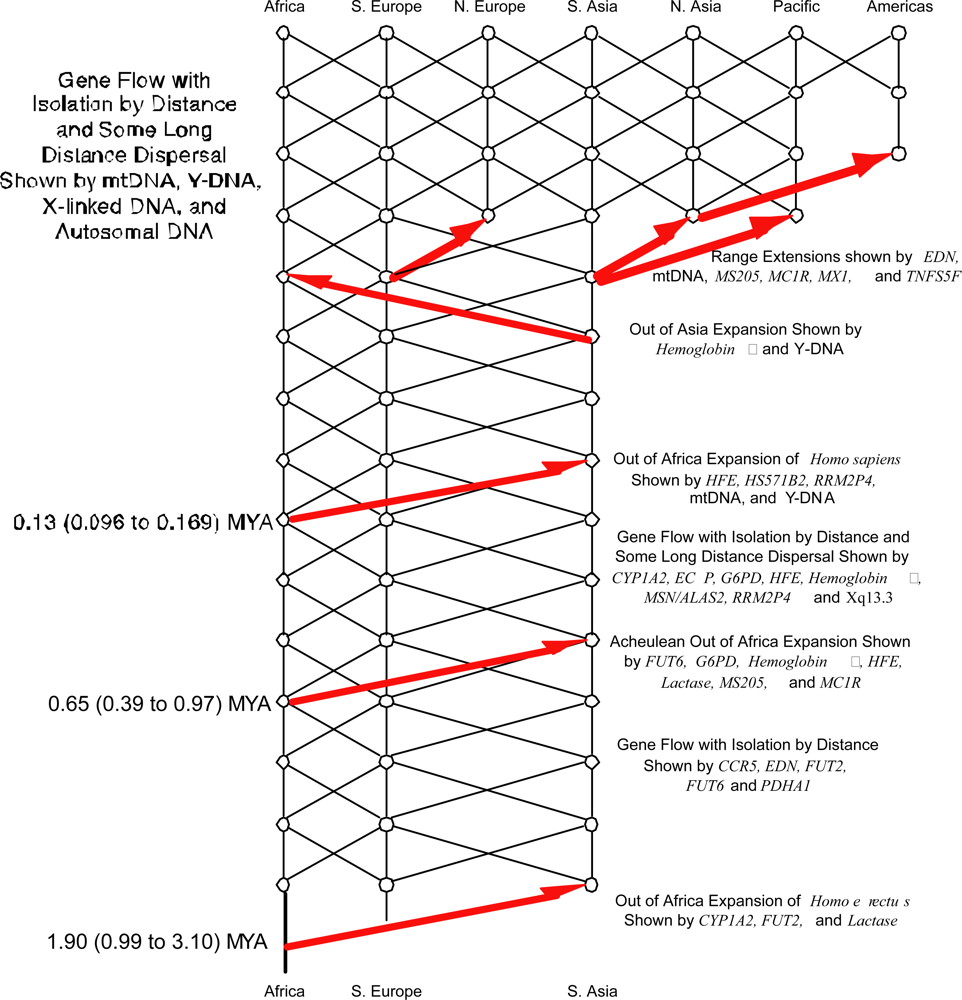
3.2. Nested Clade Phylogeographic Analysis of Human Evolution
3.3. Nested Clade Phylogeographic Analysis in Conservation Biology
4. Species Identification
5. Conclusions
Acknowledgments
References and Notes
- Hennig, W. Phylogenetic SYSTEMATICS. Annu. Rev. Entomol 1965, 10, 97–116. [Google Scholar]
- Woolley, SM; Posada, D; Crandall, KA. A Comparison of phylogenetic network methods using computer simulation. PLoS ONE 2008, 3, e1913. [Google Scholar]
- Templeton, AR. Population Genetics and Microevolutionary Theory; John Wiley & Sons: Hoboken, NJ, USA, 2006; p. 705. [Google Scholar]
- Huang, QQ; Morrison, AC; Boerwinkle, E. Linkage disequilibrium structure and its impact on the localization of a candidate functional mutation. Genet. Epidemiol 2001, 21, S620–S625. [Google Scholar]
- Templeton, AR; Maxwell, T; Posada, D; Stengard, JH; Boerwinkle, E; Sing, CF. Tree scanning: A method for using haplotype trees In genotype/phenotype association studies. Genetics 2005, 169, 441–453. [Google Scholar]
- Maruyama, K; Schoor, KD; Hartl, DL. Identification of nucleotide substitutions necessary for trans-activation of mariner transposable elements in Drosophila: Analysis of naturally occurring elements. Genetics 1991, 128, 777–784. [Google Scholar]
- Keavney, B; McKenzie, CA; Connell, JMC; Julier, C; Ratcliffe, PJ; Sobel, E; Lathrop, M; Farrall, M. Measured haplotype analysis of the Angiotensin-I Converting Enzyme gene. Hum. Mol. Genet 1998, 7, 1745–1751. [Google Scholar]
- Templeton, AR; Boerwinkle, E; Sing, CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. I. Basic theory and an analysis of Alcohol Dehydrogenase activity in Drosophila. Genetics 1987, 117, 343–351. [Google Scholar]
- Templeton, AR; Sing, CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. IV. Nested analyses with cladogram uncertainty and recombination. Genetics 1993, 134, 659–669. [Google Scholar]
- Templeton, AR. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping or DNA sequencing. V. Analysis of case/control sampling designs: Alzheimer's disease and the Apoprotein E locus. Genetics 1995, 140, 403–409. [Google Scholar]
- Prum, B; Guilloud-Bataille, M; Clerget-Darpoux, F. On the use of χ2 tests for nested categoried data. Annals Hum. Genet 1990, 54, 315–320. [Google Scholar]
- Rosenberg, S; Templeton, AR; Feigin, PD; Lancet, D; Beckmann, JS; Selig, S; Hamer, DH; Skorecki, K. The association of DNA sequence variation at the MAOA genetic locus with quantitative behavioural traits in normal males. Hum. Genet 2006, 120, 447–459. [Google Scholar]
- Templeton, AR; Sing, CF; Kessling, A; Humphries, S. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. II. The analysis of natural populations. Genetics 1988, 120, 1145–1154. [Google Scholar]
- Lobos, EA; Todd, RD. Cladistic analysis of disease association with tyrosine hydroxylase-application to manic-depressive disease and alcoholism. Am. J. Med. Genet 1997, 74, 289–295. [Google Scholar]
- Kittles, RA; Long, JC; Bergen, AW; Eggert, M; Virkkune, M; Linnoila, M; Goldman, D. Cladistic association analysis of Y chromosome effects on alcohol dependence and related personality traits. Proc. Natl. Acad. Sci. USA 1999, 96, 4204–4209. [Google Scholar]
- Hallman, DM; Visvikis, S; Steinmetz, J; Boerwinkle, E. The effect of variation in the apolipoprotein B gene on plasmid lipid and apolipoprotein B levels. I. A likelihood-based approach to cladistic analysis. Ann. Hum. Genet 1994, 58, 35–64. [Google Scholar]
- Seltman, H; Roeder, K; Devlin, B. Transmission/disequilibrium test meets measured haplotype analysis: Family-based association analysis guided by evolution of haplotypes. Am. J. Hum. Genet 2001, 68, 1250–1263. [Google Scholar]
- Hoehe, MR; Kopke, K; Wendel, B; Rohde, K; Flachmeier, C; Kidd, KK; Berrettini, WH; Church, GM. Sequence variability and candidate gene analysis in complex disease: Association of mu opioid receptor gene variation with substance dependence. Hum. Mol. Genet 2000, 9, 2895–2908. [Google Scholar]
- Morris, AP; Whittaker, JC; Balding, DJ. Fine-scale mapping of disease loci via shattered coalescent modeling of genealogies. Am. J. Hum. Genet 2002, 70, 686–707. [Google Scholar]
- Molitor, J; Marjoram, P; Thomas, D. Application of Bayesian spatial statistical methods to analysis of haplotypes effects and gene mapping. Genet. Epidemiol 2003, 25, 95–105. [Google Scholar]
- Thomas, DC; Stram, DO; Conti, D; Molitor, J; Marjoram, P. Bayesian spatial modeling of haplotype associations. Hum. Hered 2003, 56, 32–40. [Google Scholar]
- Tzeng, JY; Devlin, B; Wasserman, L; Roeder, K. On the identification of disease mutations by the analysis of haplotype similarity and goodness of fit. Am. J. Hum. Genet 2003, 72, 891–902. [Google Scholar]
- Durrant, C; Zondervan, KT; Cardon, LR; Hunt, S; Deloukas, P; Morris, AP. Linkage disequilibrium mapping via cladistic analysis of single-nucleotide polymorphism haplotypes. Am. J. Hum. Genet 2004, 75, 35–43. [Google Scholar]
- Hagenblad, J; Tang, C; Molitor, J; Werner, J; Zhao, K; Zheng, H; Marjoram, P; Weigel, D; Nordborg, M. Haplotype structure and phenotypic associations in the chromosomal regions surrounding two arabidopsis thaliana flowering time loci. Genetics 2004, 168, 1627–1638. [Google Scholar]
- Katzov, H; Bennet, AM; Kehoe, P; Wiman, B; Gatz, M; Blennow, K; Lenhard, B; Pedersen, NL; de Faire, U; Prince, JA. A cladistic model of ACE sequence variation with implications for myocardial infarction, Alzheimer disease and obesity. Hum. Mol. Genet 2004, 13, 2647–2657. [Google Scholar]
- Rodriguez, S; Gaunt, TR; Chen, XH; Gu, D; Hawe, E; Miller, GJ; Humphries, SE; Day, INM. Haplotypic analyses of the IGF2-INS-TH gene cluster in relation to cardiovascular risk traits. Hum. Mol. Genet 2004, 13, 715–725. [Google Scholar]
- Seltman, H; Roeder, K; Devlin, B. Evolutionary-based association analysis using haplotype data. Genet. Epidemiol 2003, 25, 48–58. [Google Scholar]
- Clark, TG; de Iorio, M; Griffiths, RC; Farrall, M. Finding associations in dense genetic maps: A genetic algorithm approach. Hum. Hered 2005, 60, 97–108. [Google Scholar]
- Tzeng, JY. Evolutionary-based grouping of haplotypes in association analysis. Genet. Epidemiol 2005, 28, 220–231. [Google Scholar]
- Yu, K; Xu, J; Rao, DC; Province, M. Using tree-based recursive partitioning methods to group haplotypes for increased power in association studies. Annals Hum. Genet 2005, 69, 577–589. [Google Scholar]
- Zollner, S; Pritchard, JK. Coalescent-based association mapping and fine mapping of complex trait loci. Genetics 2005, 169, 1071–1092. [Google Scholar]
- Minichiello, MJ; Durbin, R. Mapping trait loci by use of inferred ancestral recombination graphs. Am. J. Hum. Genet 2006, 79, 910–922. [Google Scholar]
- Morris, AP. A flexible Bayesian framework for modeling haplotype association with disease, allowing for dominance effects of the underlying causative variants. Am. J. Hum. Genet 2006, 79, 679–694. [Google Scholar]
- Tzeng, J-Y; Wang, C-H; Kao, J-T; Hsiao, CK. Regression-based association analysis with clustered haplotypes through use of genotypes. Am. J. Hum. Genet 2006, 78, 231–242. [Google Scholar]
- Waldron, ERB; Whittaker, JC; Balding, DJ. Fine mapping of disease genes via haplotype clustering. Genet. Epidemiol 2006, 30, 170–179. [Google Scholar]
- Liu, J; Papasian, C; Deng, H-W. Incorporating single-locus tests into haplotype cladistic analysis in case-control studies. PLoS Genet 2007, 3, e46. [Google Scholar]
- Sjolander, A; Hossjer, O; Hartman, LW; Humphreys, K. Fine mapping of disease genes using tagging SNPs. Annals Hum. Genet 2007, 71, 815–827. [Google Scholar]
- Tachmazidou, I; Verzilli, CJ; Iorio, MD. Genetic association mapping via evolution-based clustering of haplotypes. PLoS Genet 2007, 3, e111. [Google Scholar]
- Knight, J; Curtis, D; Sham, PC. CLUMPHAP: A simple tool for performing haplotype-based association analysis. Genet. Epidemiol 2008, 32, 539–545. [Google Scholar]
- Larribe, F; Lessard, S. A composite-conditional-likelihood approach for gene mapping based on linkage disequilibrium in windows of marker loci. Stat Appl Genet Mol Biol 2008, 7. [Google Scholar]
- Besenbacher, S; Mailund, T; Schierup, MH. Local phylogeny mapping of quantitative traits: Higher accuracy and better ranking than single-marker association in genomewide scans. Genetics 2009, 181, 747–753. [Google Scholar]
- Liu, Y; Li, Y-J; Satten, GA; Allen, AS; Tzeng, J-Y. A regression-based association test for case-control studies that uses inferred ancestral haplotype similarity. Annals Hum. Genet 2009, 73, 520–526. [Google Scholar]
- Tapper, W; Gibson, J; Morton, NE; Collins, A. A comparison of methods to detect recombination hotspots. Hum. Hered 2008, 66, 157–169. [Google Scholar]
- Templeton, AR; Routman, E; Phillips, C. Separating population structure from population history: A cladistic analysis of the geographical distribution of mitochondrial DNA haplotypes in the tiger salamander Ambystoma tigrinum. Genetics 1995, 140, 767–782. [Google Scholar]
- Templeton, AR. Nested clade analysis: An extensively validated method for strong phylogeographic inference. Mol. Ecol 2008, 17, 1877–1880. [Google Scholar]
- Templeton, AR. Out of Africa again and again. Nature 2002, 416, 45–51. [Google Scholar]
- Templeton, AR. Why does a method that fails continue to be used: The answer. Evolution 2009, 63, 807–812. [Google Scholar]
- Templeton, AR. A maximum likelihood framework for cross validation of phylogeographic hypotheses. In Evolutionary Theory and Processes: Modern Horizons; Wasser, SP, Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp. 209–230. [Google Scholar]
- Templeton, AR. Statistical phylogeography: Methods of evaluating and minimizing inference errors. Mol. Ecol 2004, 13, 789–809. [Google Scholar]
- Fagundes, NJR; Ray, N; Beaumont, M; Neuenschwander, S; Salzano, FM; Bonatto, SL; Excoffier, L. Statistical evaluation of alternative models of human evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 17614–17619. [Google Scholar]
- Templeton, AR. Statistical hypothesis testing in intraspecific phylogeography: Nested clade phylogeographical analysis vs. approximate Bayesian computation. Mol. Ecol 2009, 18, 319–331. [Google Scholar]
- Templeton, AR. Haplotype trees and modern human origins. Yearb. Phys. Anthropol 2005, 48, 33–59. [Google Scholar]
- Templeton, AR. Perspective: Genetics and recent human evolution. Evolution 2007, 61, 1507–1519. [Google Scholar]
- Templeton, AR. Gene flow, haplotype patterns and modern human origins. In Encyclopedia of Life Sciences; Wiley: Chichester, UK, 2007. [Google Scholar]
- Templeton, AR. Population biology and population genetics of Pleistocene Hominins. In Handbook of Palaeoanthropology; Henke, W, Tattersall, I, Eds.; Springer-Verlag: Berlin, German, 2007; Volume 3, pp. 1825–1859. [Google Scholar]
- Moritz, C. Defining ‘Evolutionary Significant Units’ for conservation. Tr. Evol. Ecol 1994, 9, 373–375. [Google Scholar]
- Roca, AL; Georgiadis, N; O'Brien, SJ. Cytonuclear genomic dissociation in African elephant species. Nat. Gen 2005, 37, 96–100. [Google Scholar]
- Templeton, AR; Georgiadis, NJ. A landscape approach to conservation genetics: Conserving evolutionary processes in the African Bovidae. In Conservation Genetics: Case Histories From Nature; Avise, JC, Hamrick, JL, Eds.; Chapman & Hall: New York, NY, USA, 1996; pp. 398–430. [Google Scholar]
- Hull, DL. The ideal species concept--and why we can't get it. In Species: The Units of Biodiversity; Claridge, MF, Dawah, HA, Wilson, MR, Eds.; Chapman & Hall: London, UK, 1997; Chaper 18pp. 357–380. [Google Scholar]
- Templeton, AR. The meaning of species and speciation: A genetic perspective. In Speciation and Its Consequences; Otte, D, Endler, JA, Eds.; Sinauer: Sunderland, MA, USA, 1989; pp. 3–27. [Google Scholar]
- Templeton, AR. Using phylogeographic analyses of gene trees to test species status and processes. Mol. Ecol 2001, 10, 779–791. [Google Scholar]
- Ebersberger, I; Galgoczy, P; Taudien, S; Taenzer, S; Platzer, M; von Haeseler, A. Mapping human genetic ancestry. Mol. Biol. Evol 2007, 24, 2266–2276. [Google Scholar]
- Roeder, K; Luca, D. Searching for disease susceptibility variants in structured populations. Genomics 2009, 93, 1–4. [Google Scholar]
- Templeton, AR. Using gene trees to infer species from testable null hypothesis: Cohesion species in the Spalax ehrenbergi complex. In Evolutionary Theory and Processes: Modern Perspectives, Papers in Honour of Eviatar Nevo; Wasser, SP, Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1999; pp. 171–192. [Google Scholar]
- Wahrman, J; Richler, C; Gamperl, R; Nevo, E. Revisiting Spalax: Mitotic and meiotic chromosome variability. Isr. J. Zool 1985, 33, 15–38. [Google Scholar]



© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Templeton, A.R. The Diverse Applications of Cladistic Analysis of Molecular Evolution, with Special Reference to Nested Clade Analysis. Int. J. Mol. Sci. 2010, 11, 124-139. https://doi.org/10.3390/ijms11010124
Templeton AR. The Diverse Applications of Cladistic Analysis of Molecular Evolution, with Special Reference to Nested Clade Analysis. International Journal of Molecular Sciences. 2010; 11(1):124-139. https://doi.org/10.3390/ijms11010124
Chicago/Turabian StyleTempleton, Alan R. 2010. "The Diverse Applications of Cladistic Analysis of Molecular Evolution, with Special Reference to Nested Clade Analysis" International Journal of Molecular Sciences 11, no. 1: 124-139. https://doi.org/10.3390/ijms11010124