Involvement of Glutamate Transporter-1 in Neuroprotection against Global Brain Ischemia-Reperfusion Injury Induced by Postconditioning in Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
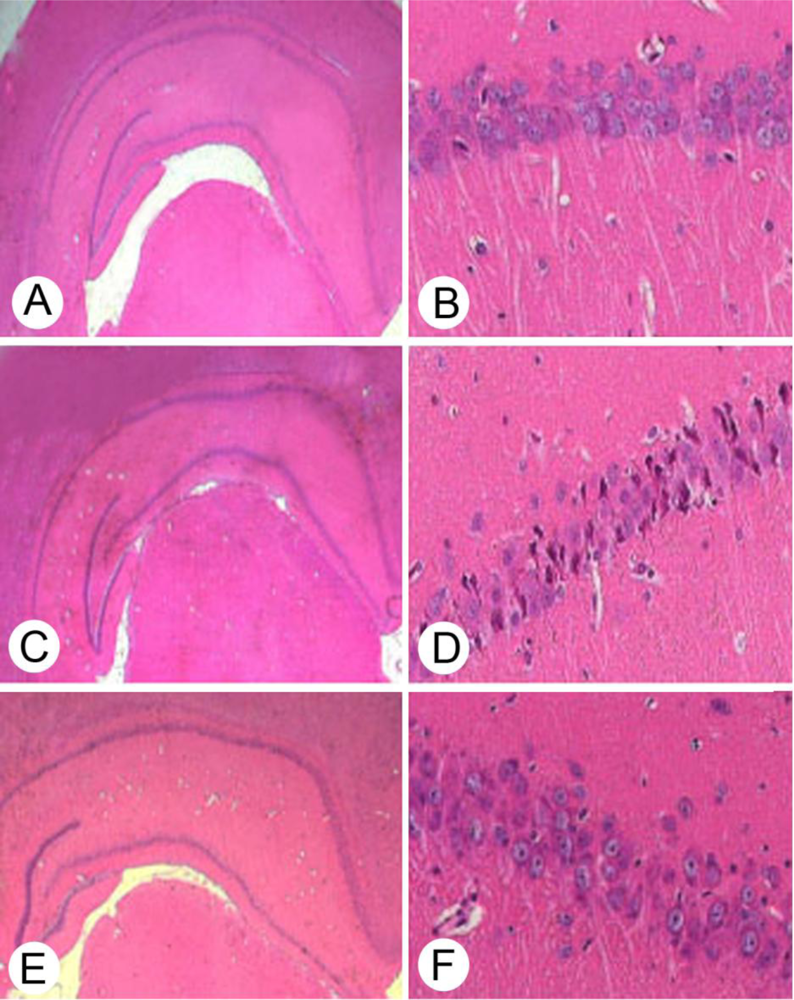
2.1. Neuropathological Evaluation of Hippocampal CA1 by Hematoxylin/Erosin Staining
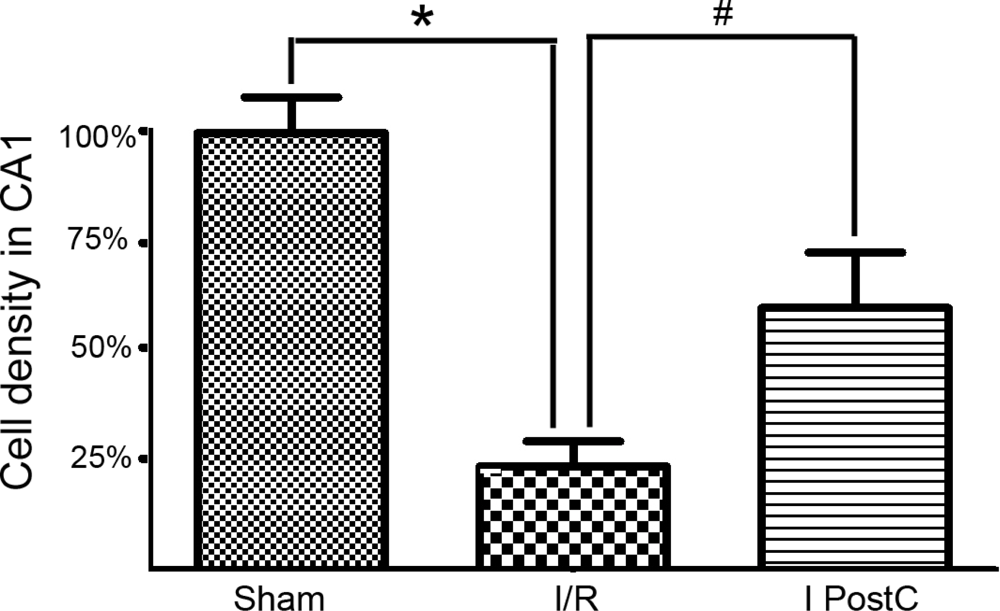
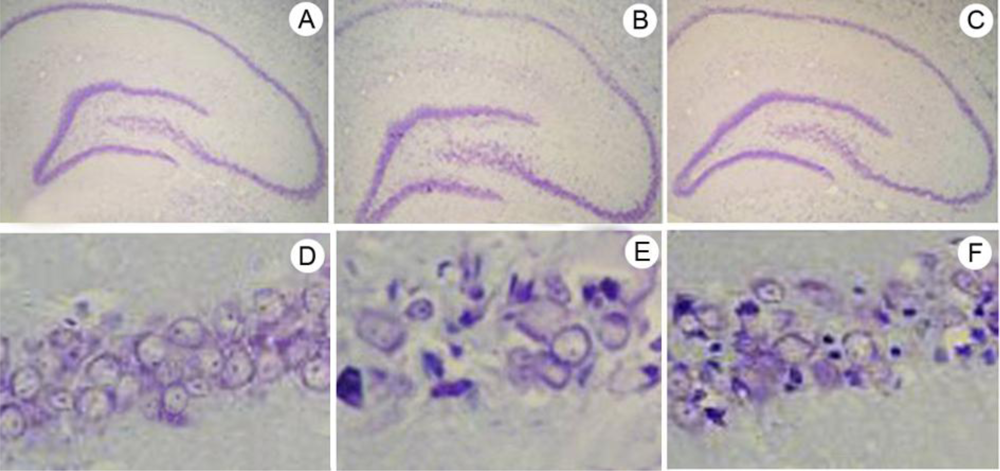
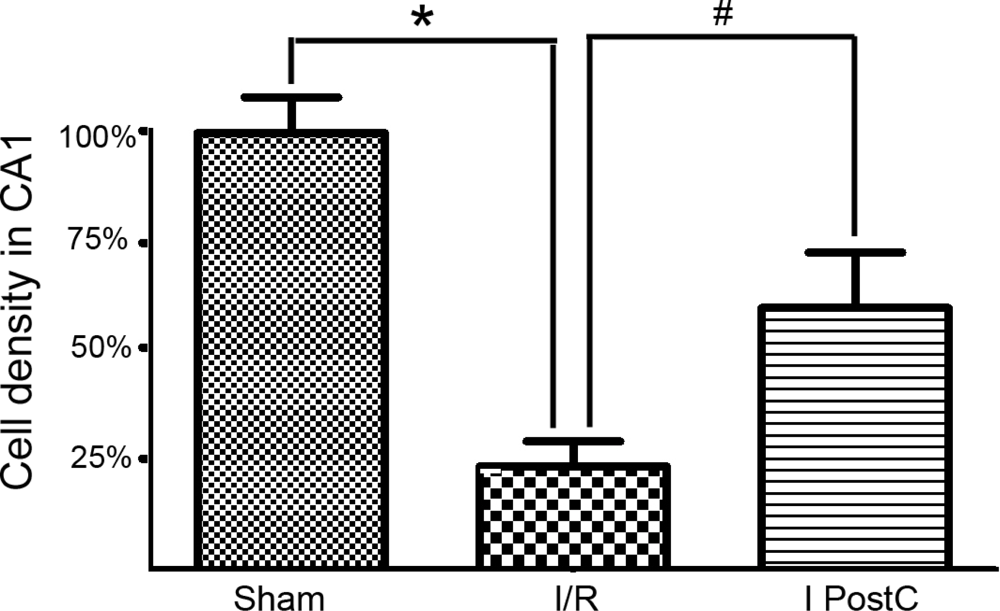
2.2. Neuron Counts in Hippocampal CA1, as Determined by Cresyl Violet Staining
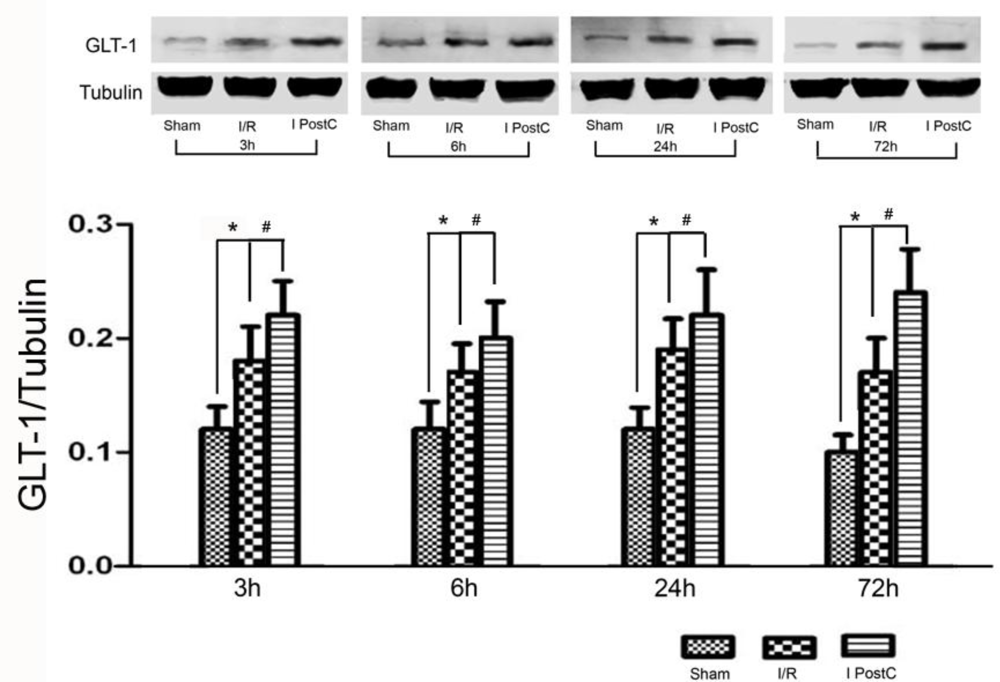
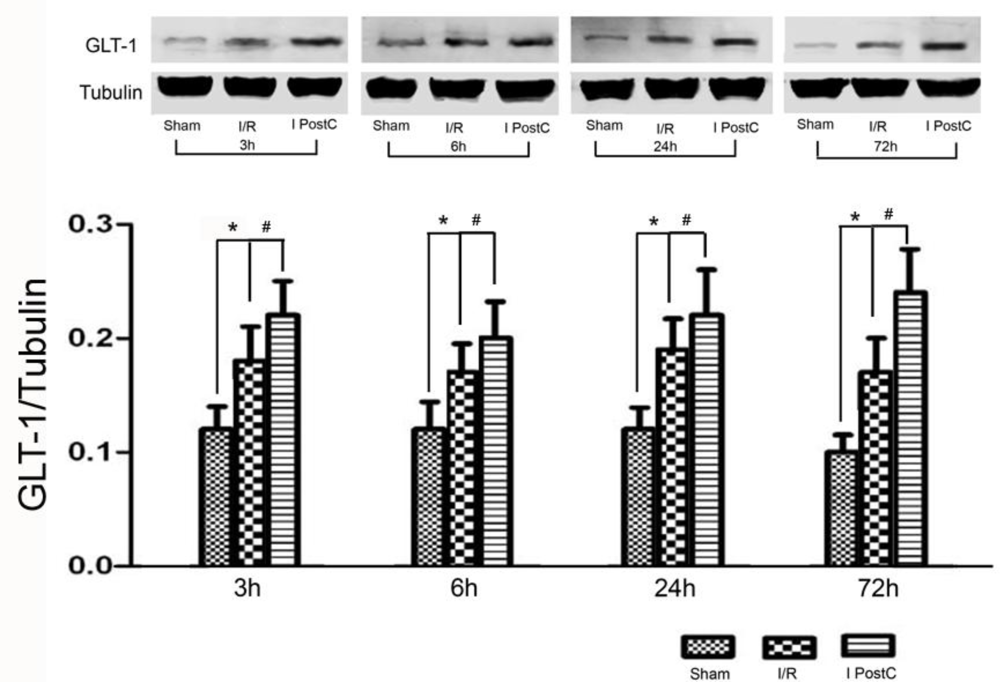
2.3. Expression of GLT-1
2.4. Discussion
3. Experimental Section
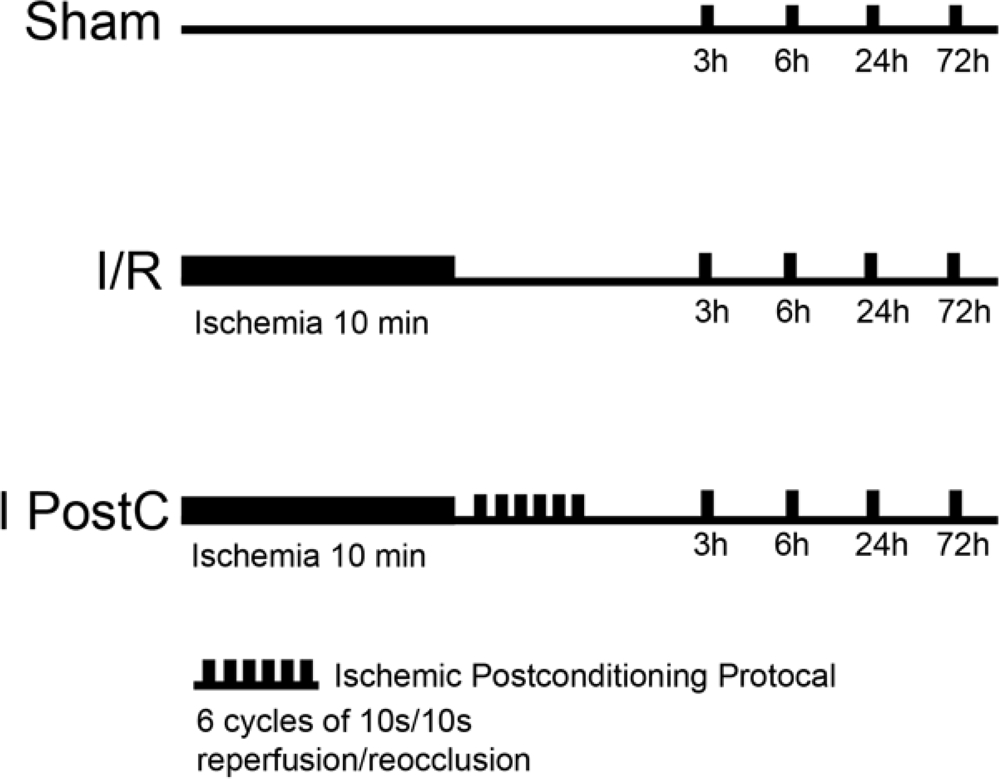
3.1. Animal Groups
3.2. Animal Model and Surgical Procedure
3.3. Histological Assessment
3.4. Western Blotting
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Mantz, J; Degos, V; Laigle, C. Recent advances in pharmacologic neuroprotection. Eur. J. Anaesthesiol 2010, 27, 6–10. [Google Scholar]
- Xing, BZ; Chen, H; Zhang, M; Zhao, DM; Jiang, R; Liu, XH; Zhang, SM. Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke 2008, 39, 2362–2369. [Google Scholar]
- Wang, JY; Shen, J; Gao, Q; Ye, ZG; Yang, SY; Liang, HW; Bruce, IC; Luo, BY; Xia, Q. Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke 2008, 39, 983–990. [Google Scholar]
- Ren, CC; Yan, ZM; Wei, DT; Gao, XW; Chen, XY; Zhao, H. Limb remote ischemic postconditioning protects against focal ischemia in rats. Brain Res 2009, 1288, 88–94. [Google Scholar]
- Taskapilioglu, MO; Alkan, T; Goren, B; Tureyen, K; Sahin, S; Taskapilioglu, O; Korfali, E. Neuronal protective effects of focal ischemic pre- and/or postconditioning on the model of transient focal cerebral ischemia in rats. J. Clin. Neurosci 2009, 16, 693–697. [Google Scholar]
- Lee, SG; Su, ZZ; Emdad, L; Gupta, P; Sarkar, D; Borjabad, A; Volsky, DJ; Fisher, PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem 2008, 283, 13116–13123. [Google Scholar]
- Guo, ZH; Li, F; Wang, WZ. The mechanisms of brain ischemic insult and potential protective interventions. Neurosci. Bull 2009, 25, 139–152. [Google Scholar]
- Ozawa, S. Role of glutamate transporters in excitatory synapses in cerebellar Purkinje cells. Brain Nerve 2007, 59, 669–676. [Google Scholar]
- Rao, VL; Dogan, A; Todd, KG; Bowen, KK; Kim, BT; Rothstein, JD; Dempsey, RJ. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci 2001, 21, 1876–1883. [Google Scholar]
- Gao, X; Ren, C; Zhao, H. Protective effects of ischemic postconditioning compared with gradual reperfusion or preconditioning. J. Neurosci. Res 2008, 86, 2505–2511. [Google Scholar]
- Gao, X; Zhang, H; Takahashi, T; Hsieh, J; Liao, J; Steinberg, GK; Zhao, H. The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J. Neurochem 2008, 105, 943–955. [Google Scholar]
- Nicotera, P; Bano, D. The enemy at the gates: Ca2+ entry through TRPM7 channels and anoxic neuronal death. Cell 2003, 115, 768–770. [Google Scholar]
- Candelario-Jalil, E. Injury and repair mechanisms in ischemic stroke: Considerations for the development of novel neurotherapeutics. Curr. Opin. Investig. Drugs 2009, 10, 644–654. [Google Scholar]
- Taoufik, E; Probert, L. Ischemic neuronal damage. Curr. Pharm. Des 2008, 14, 3565–3573. [Google Scholar]
- Romera, C; Hurtado, O; Mallolas, J; Pereira, MP; Morales, JR; Romera, A; Serena, J; Vivancos, J; Nombela, F; Lorenzo, P; et al. Ischemic preconditioning reveals that GLT1/EAAT2 glutamate transporter is a novel PPARgamma target gene involved in neuroprotection. J. Cereb. Blood Flow Metab 2007, 27, 1327–1338. [Google Scholar]
- Chen, JC; Hsu-Chou, H; Lu, JL; Chiang, YC; Huang, HM; Wang, HL; Wu, T; Liao, JJ; Yeh, TS. Down-regulation of the glial glutamate transporter GLT-1 in rat hippocampus and striatum and its modulation by a group III metabotropic glutamate receptor antagonist following transient global forebrain ischemia. Neuropharmacology 2005, 49, 703–714. [Google Scholar]
- Han, F; Shioda, N; Moriguchi, S; Qin, ZH; Fukunaga, K. Downregulation of glutamate transporters is associated with elevation in extracellular glutamate concentration following rat microsphere embolism. Neurosci. Lett 2008, 430, 275–280. [Google Scholar]
- Rao, VLR; Dogan, A; Rothstein, JD; Dempsey, RJ. Decreased glial glutamate transporter GLT-1 protein levels in rat brain following transient middle cerebral artery occlusion. J. Neurochem 1998, 70, S68. [Google Scholar]
- Rothstein, JD; Dykes-Hoberg, M; Pardo, CA; Bristol, LA; Jin, L; Kuncl, RW; Kanai, Y; Hediger, MA; Wang, Y; Schielke, JP; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar]
- Chu, K; Lee, ST; Sinn, DI; Ko, SY; Kim, EH; Kim, JM; Kim, SJ; Park, DK; Jung, KH; Song, EC; et al. Pharmacological Induction of Ischemic Tolerance by Glutamate Transporter-1 (EAAT2) Upregulation. Stroke 2007, 38, 177–182. [Google Scholar]
- Li, L; Lundkvist, A; Andersson, D; Wilhelmsson, U; Nagai, N; Pardo, AC; Nodin, C; Stahlberg, A; Aprico, K; Larsson, K; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab 2008, 28, 468–481. [Google Scholar]
- Vanhoutte, N; Abarca-Quinones, J; Jordan, BF; Gallez, B; Maloteaux, JM; Hermans, E. Enhanced expression of the high affinity glutamate transporter GLT-1 in C6 glioma cells delays tumour progression in rat. Exp. Neurol 2009, 218, 56–63. [Google Scholar]
- Bjornsen, LP; Eid, T; Holmseth, S; Danbolt, NC; Spencer, DD; de Lanerolle, NC. Changes in glial glutamate transporters in human epileptogenic hippocampus: Inadequate explanation for high extracellular glutamate during seizures. Neurobiol. Dis 2007, 25, 319–330. [Google Scholar]
- Yi, JH; Hazell, AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int 2006, 48, 394–403. [Google Scholar]
- Matute, C; Melone, M; Vallejo-Illarramendi, A; Conti, F. Increased expression of the astrocytic glutamate transporter GLT-1 in the prefrontal cortex of schizophrenics. Glia 2005, 49, 451–455. [Google Scholar]
- Namura, S; Maeno, H; Takami, S; Jiang, XF; Kamichi, S; Wada, K; Nagata, I. Inhibition of glial glutamate transporter GLT-1 augments brain edema after transient focal cerebral ischemia in mice. Neurosci. Lett 2002, 324, 117–120. [Google Scholar]
- Lu, Z; Zhang, W; Zhang, N; Jiang, J; Luo, Q; Qiu, Y. The expression of glutamate transporters in chest compression-induced audiogenic epilepsy: a comparative study. Neurol. Res 2008, 30, 915–919. [Google Scholar]
- Miao, Y; Zhang, W; Lin, Y; Lu, X; Qiu, Y. Neuroprotective effects of ischemic preconditioning on global brain ischemia through up-regulation of acid-sensing ion channel 2a. Int. J. Mol. Sci 2010, 11, 140–153. [Google Scholar]

© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, W.; Miao, Y.; Zhou, S.; Wang, B.; Luo, Q.; Qiu, Y. Involvement of Glutamate Transporter-1 in Neuroprotection against Global Brain Ischemia-Reperfusion Injury Induced by Postconditioning in Rats. Int. J. Mol. Sci. 2010, 11, 4407-4416. https://doi.org/10.3390/ijms11114407
Zhang W, Miao Y, Zhou S, Wang B, Luo Q, Qiu Y. Involvement of Glutamate Transporter-1 in Neuroprotection against Global Brain Ischemia-Reperfusion Injury Induced by Postconditioning in Rats. International Journal of Molecular Sciences. 2010; 11(11):4407-4416. https://doi.org/10.3390/ijms11114407
Chicago/Turabian StyleZhang, Weiqiao, Yifeng Miao, Sanquan Zhou, Baofeng Wang, Qizhong Luo, and Yongming Qiu. 2010. "Involvement of Glutamate Transporter-1 in Neuroprotection against Global Brain Ischemia-Reperfusion Injury Induced by Postconditioning in Rats" International Journal of Molecular Sciences 11, no. 11: 4407-4416. https://doi.org/10.3390/ijms11114407