Comparison of TNFα to Lipopolysaccharide as an Inflammagen to Characterize the Idiosyncratic Hepatotoxicity Potential of Drugs: Trovafloxacin as an Example

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Serum Chemistry and Histopathology for the TNFα Study
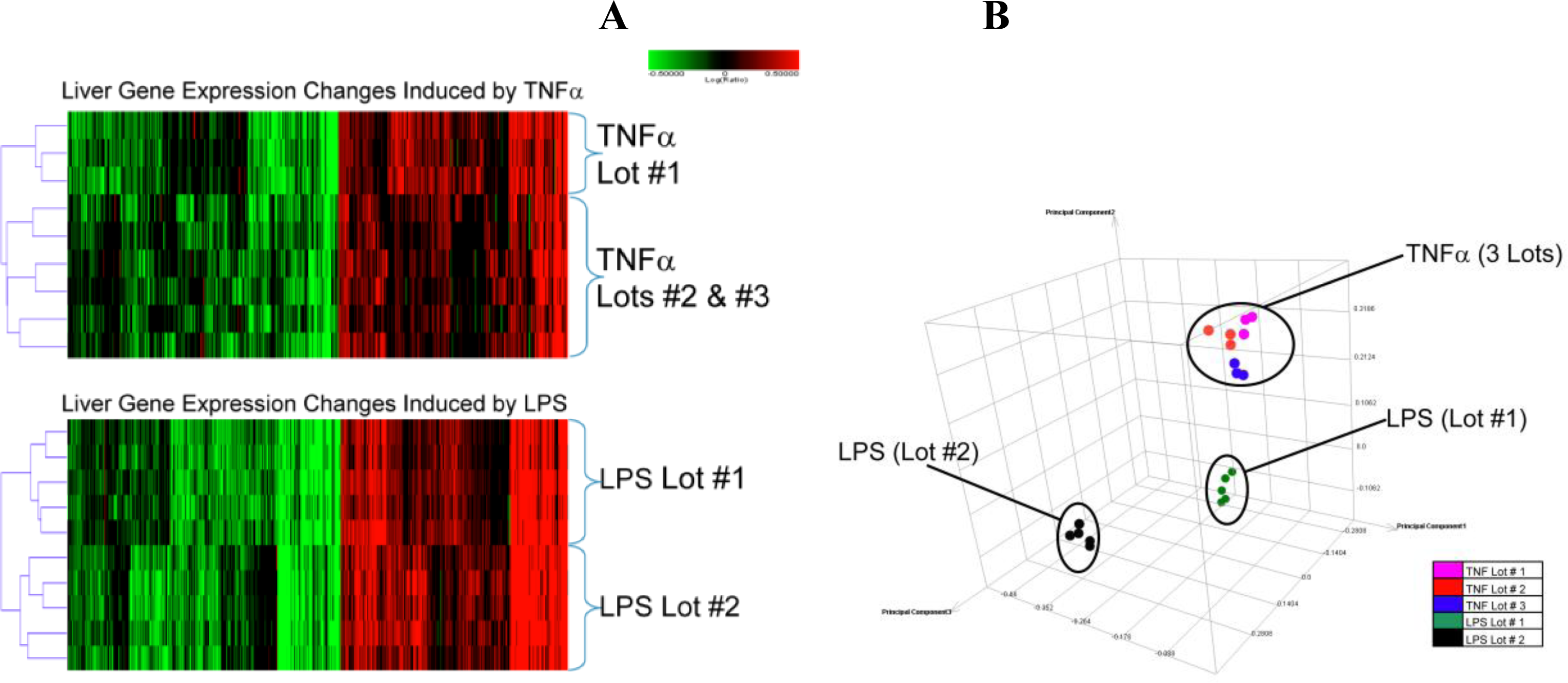
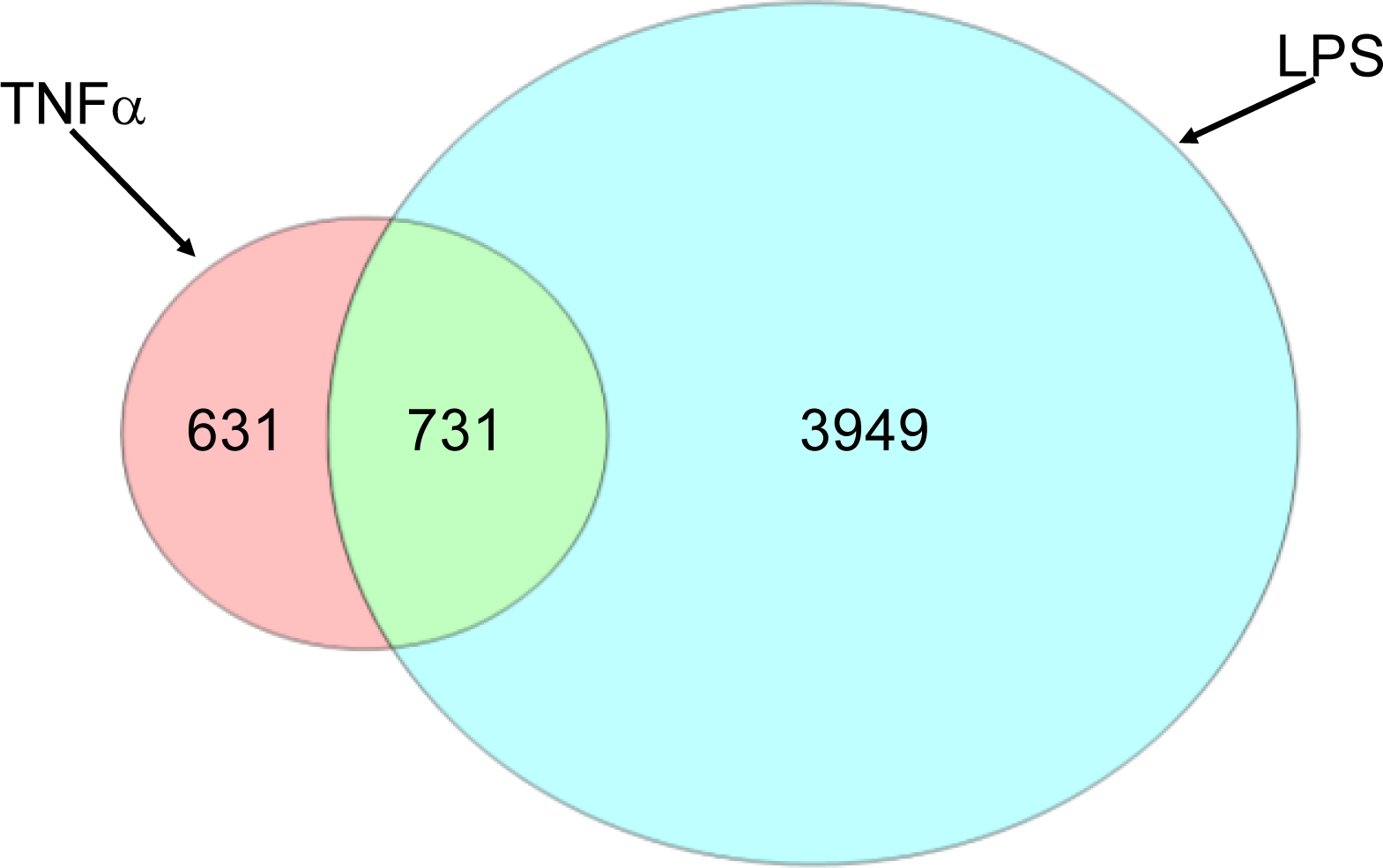
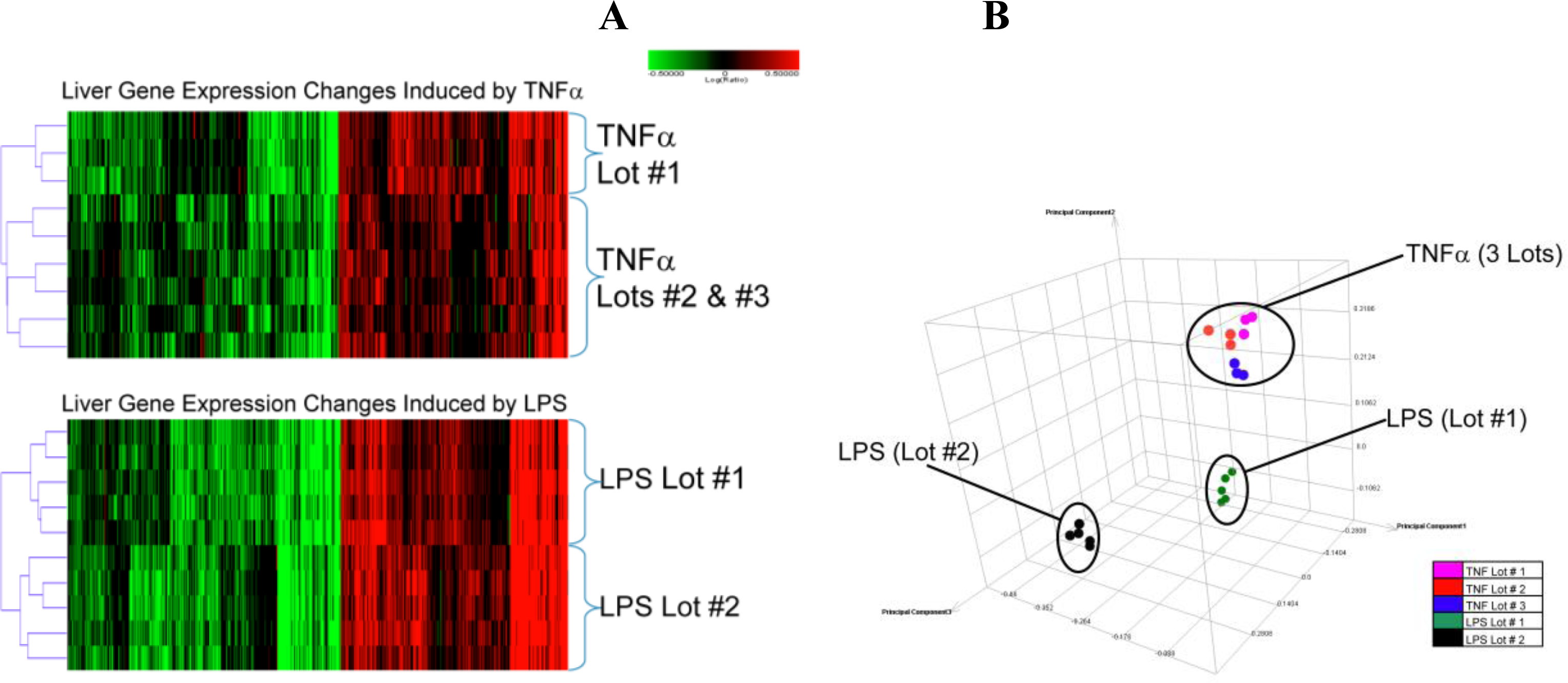
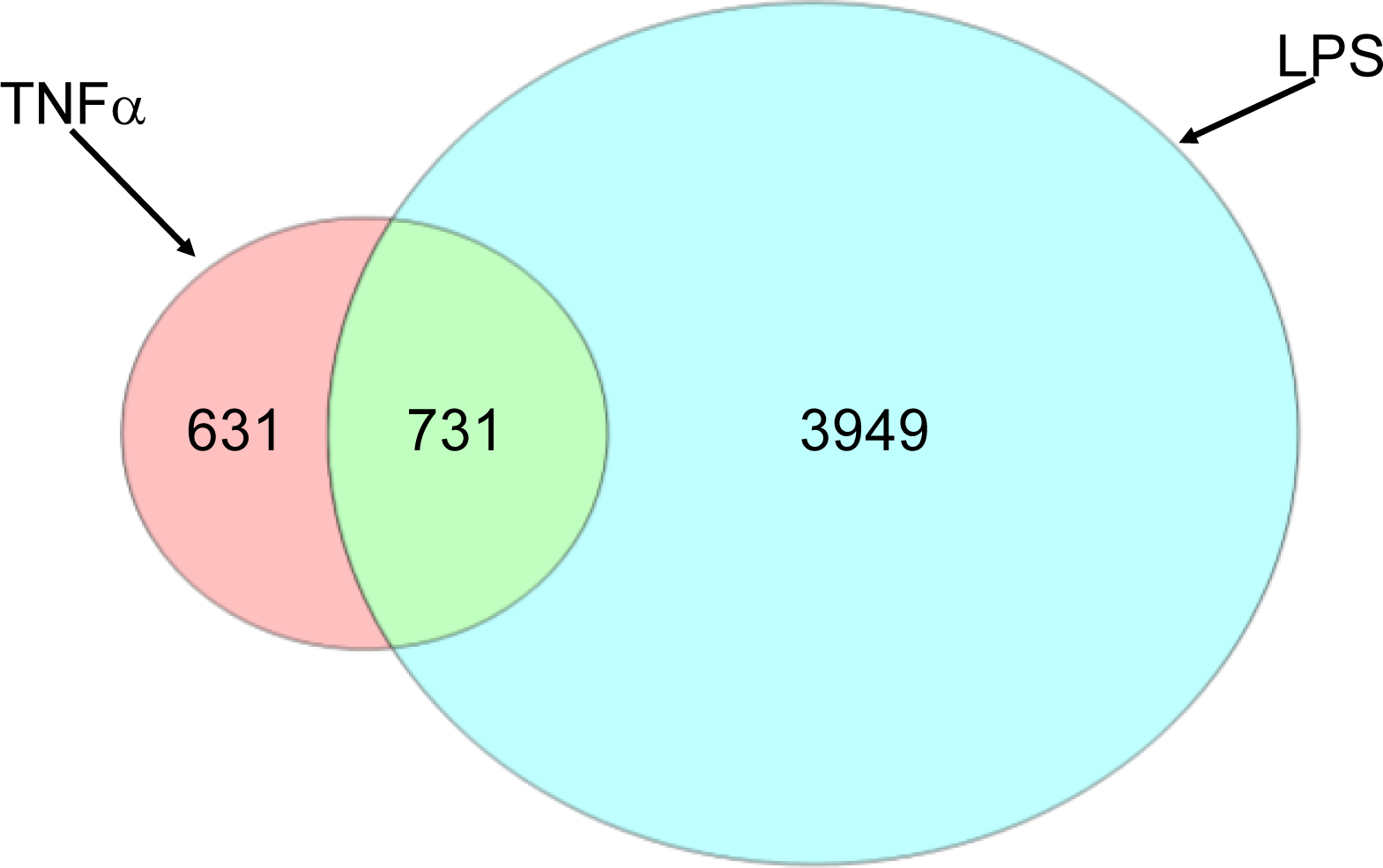
2.1.2. Comparison of Hepatic Transcriptomes Induced by LPS or TNFα
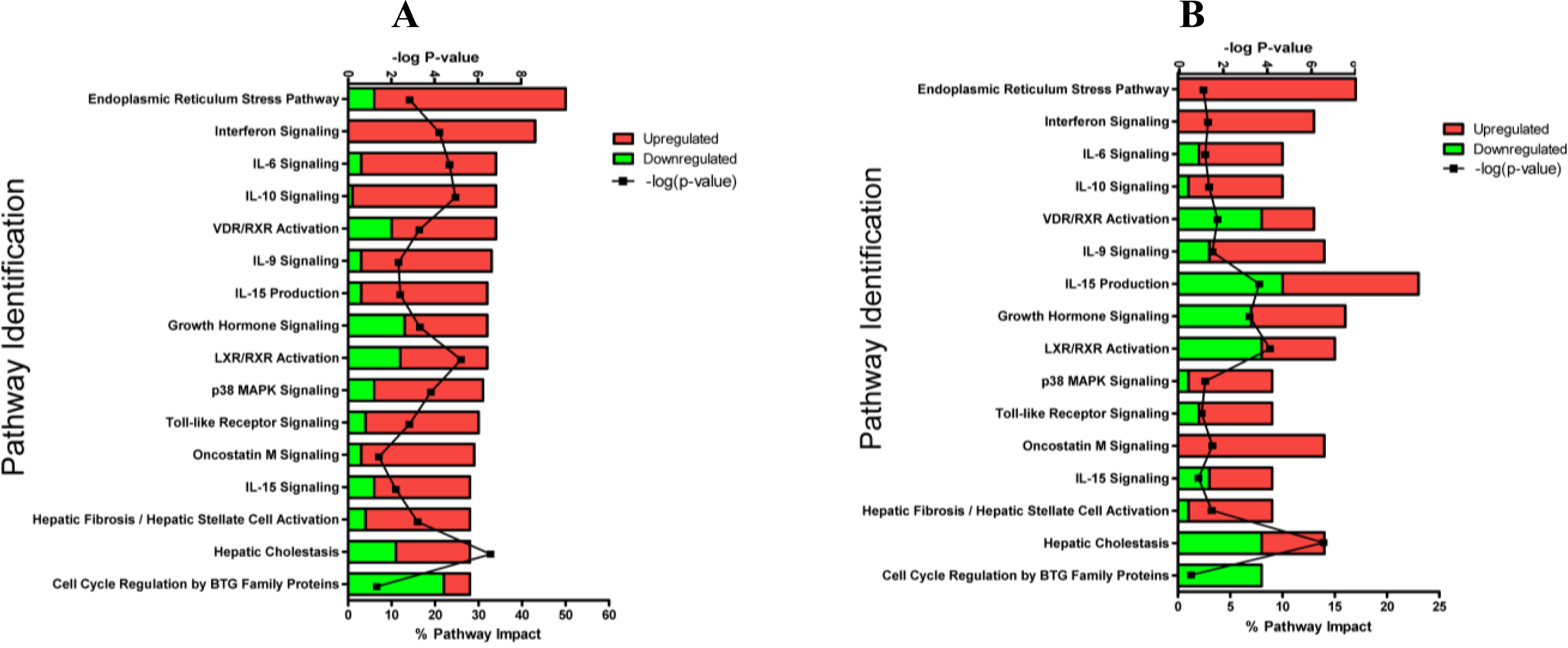
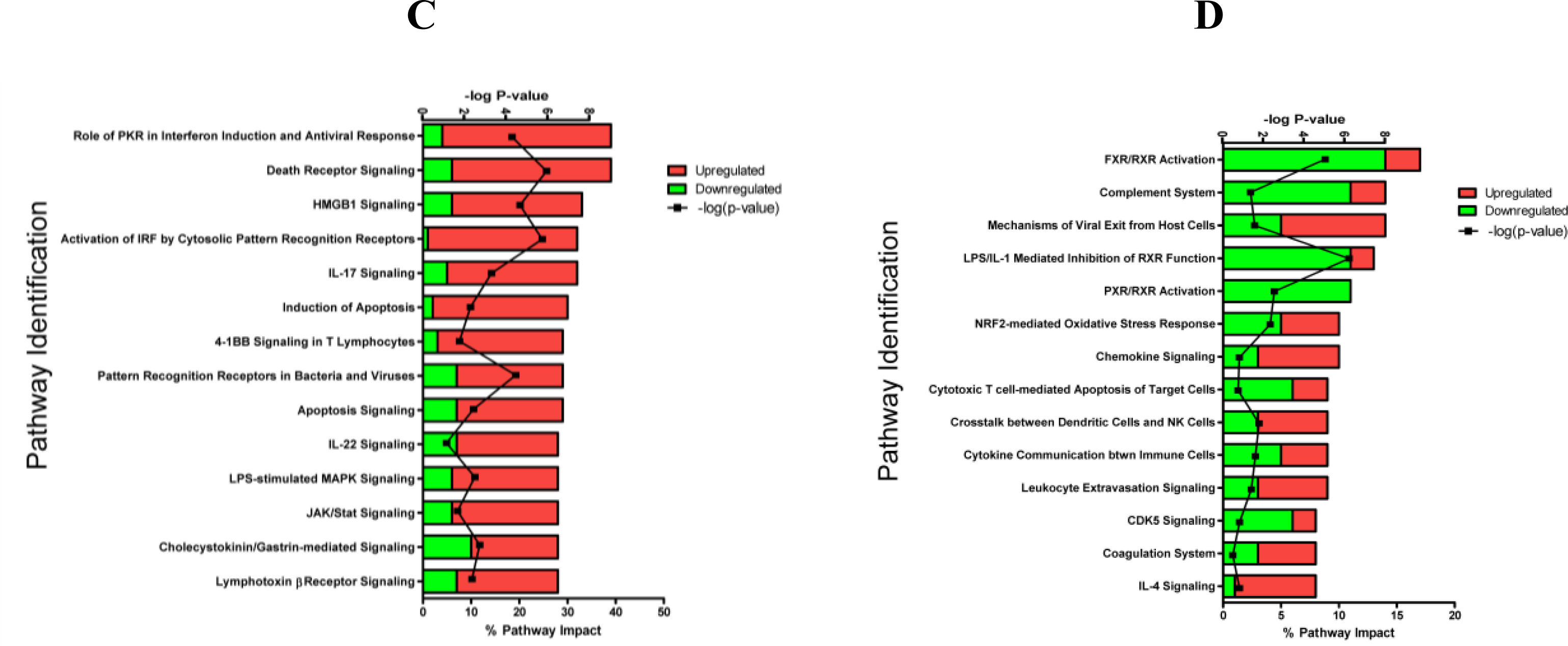
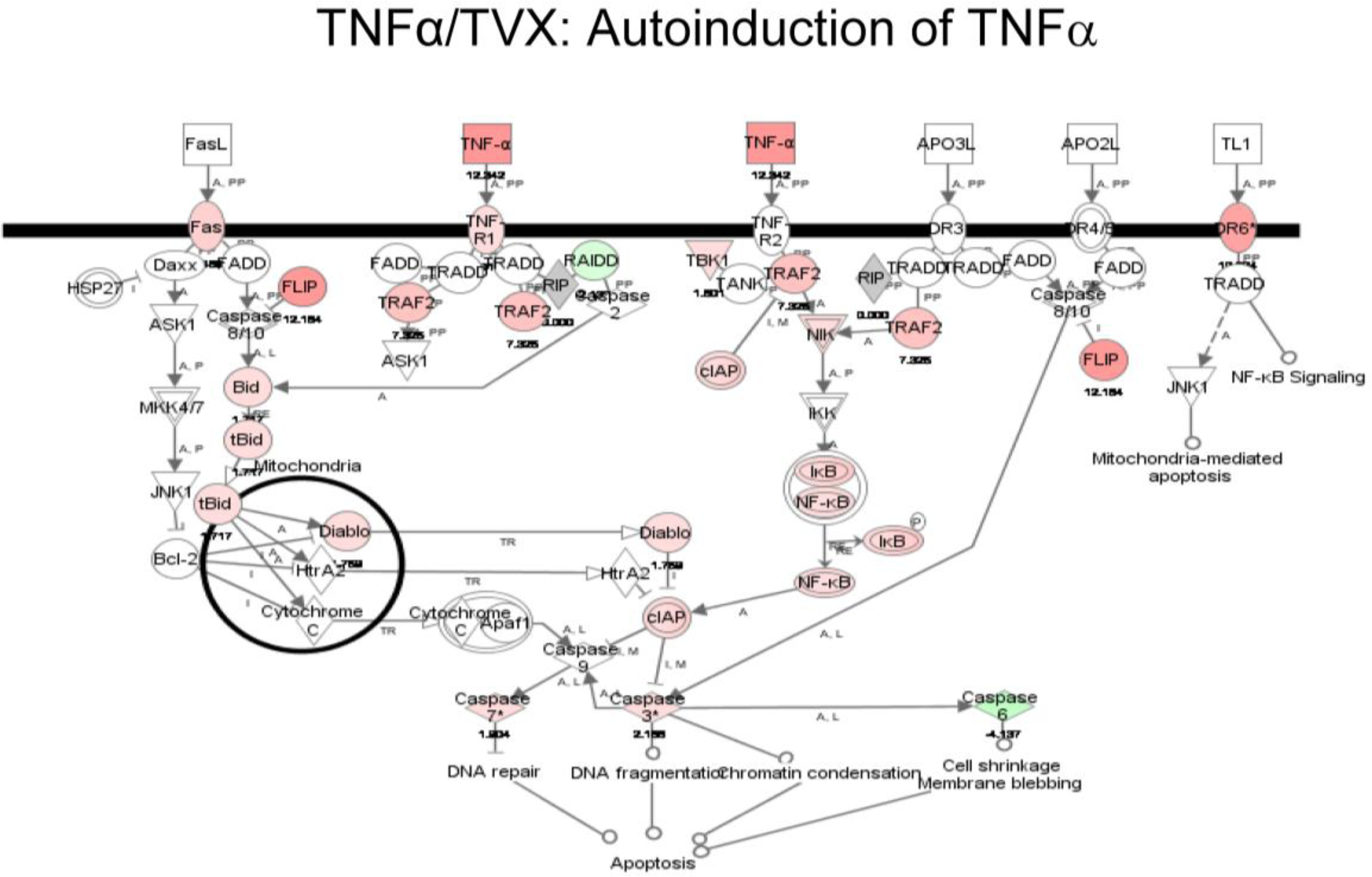
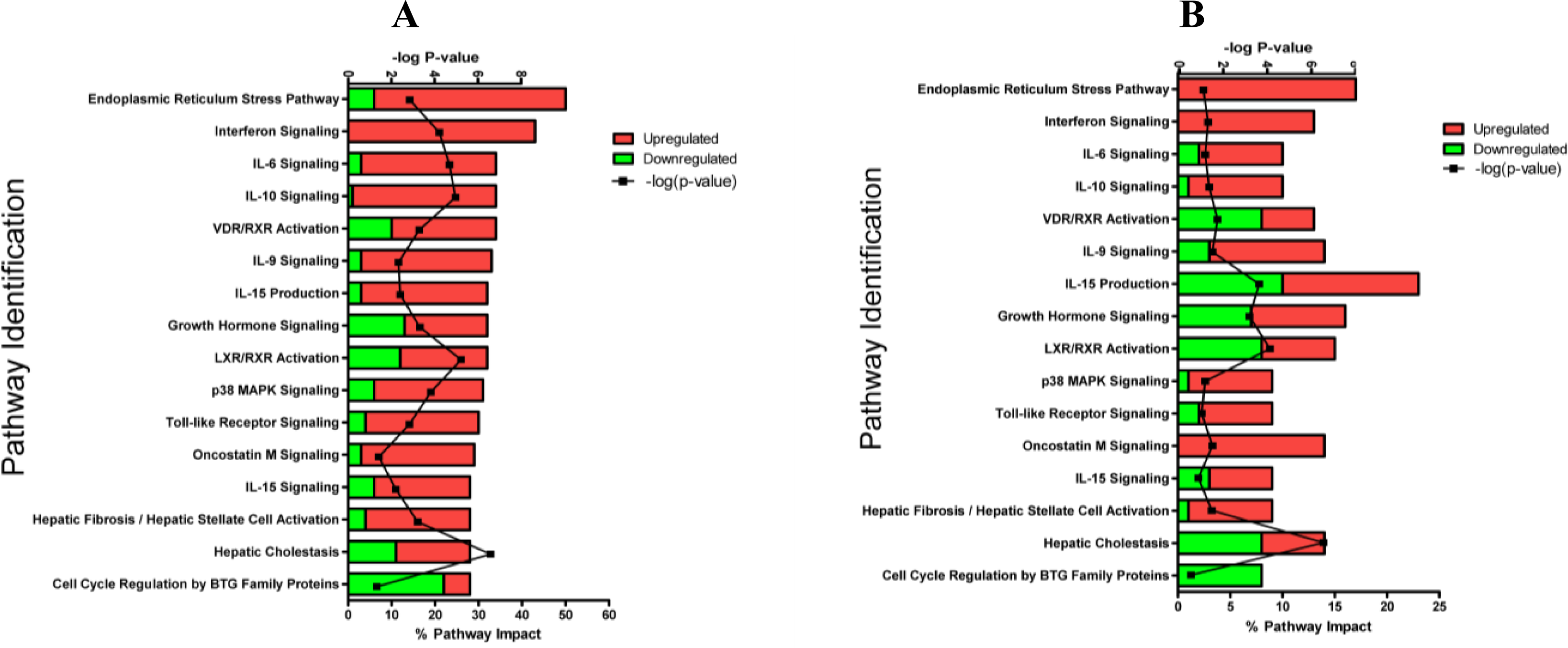
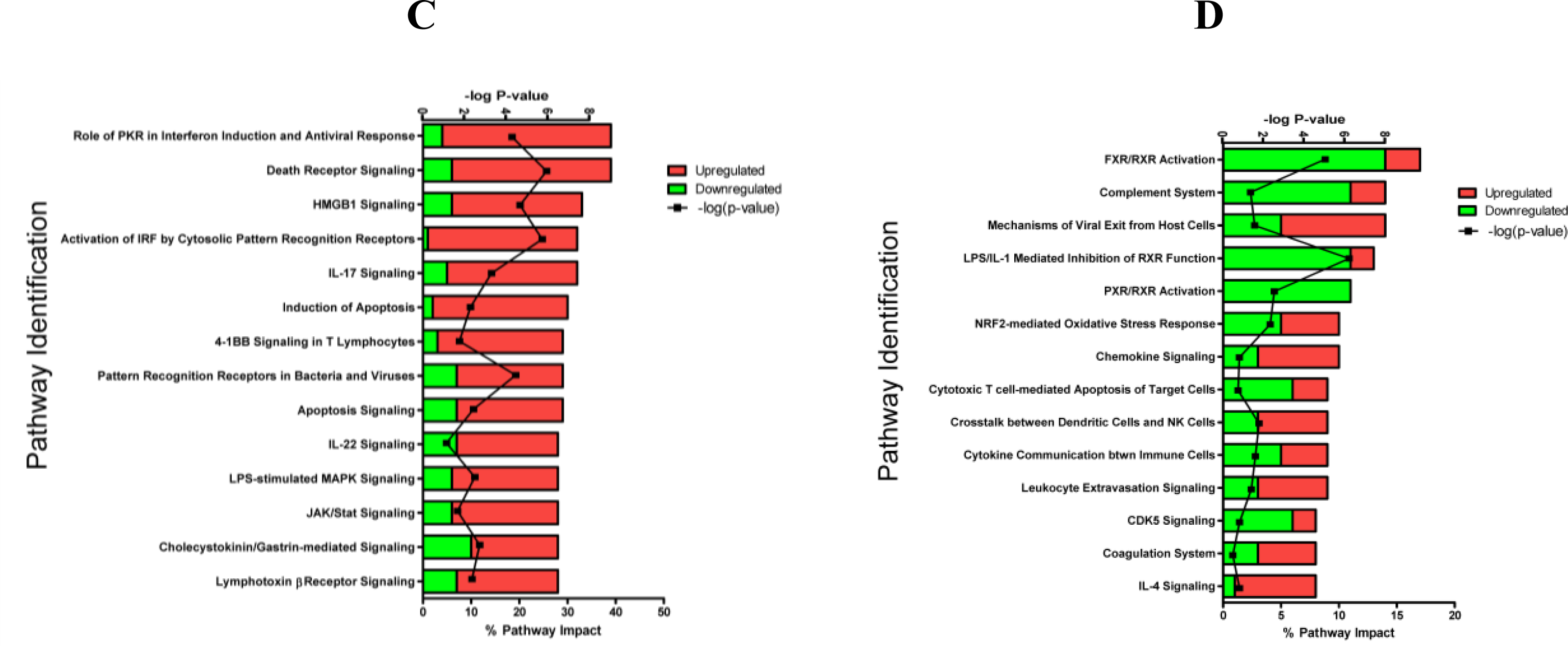
2.1.3. Classification of Biochemical Pathways Significantly Impacted by LPS or TNFα
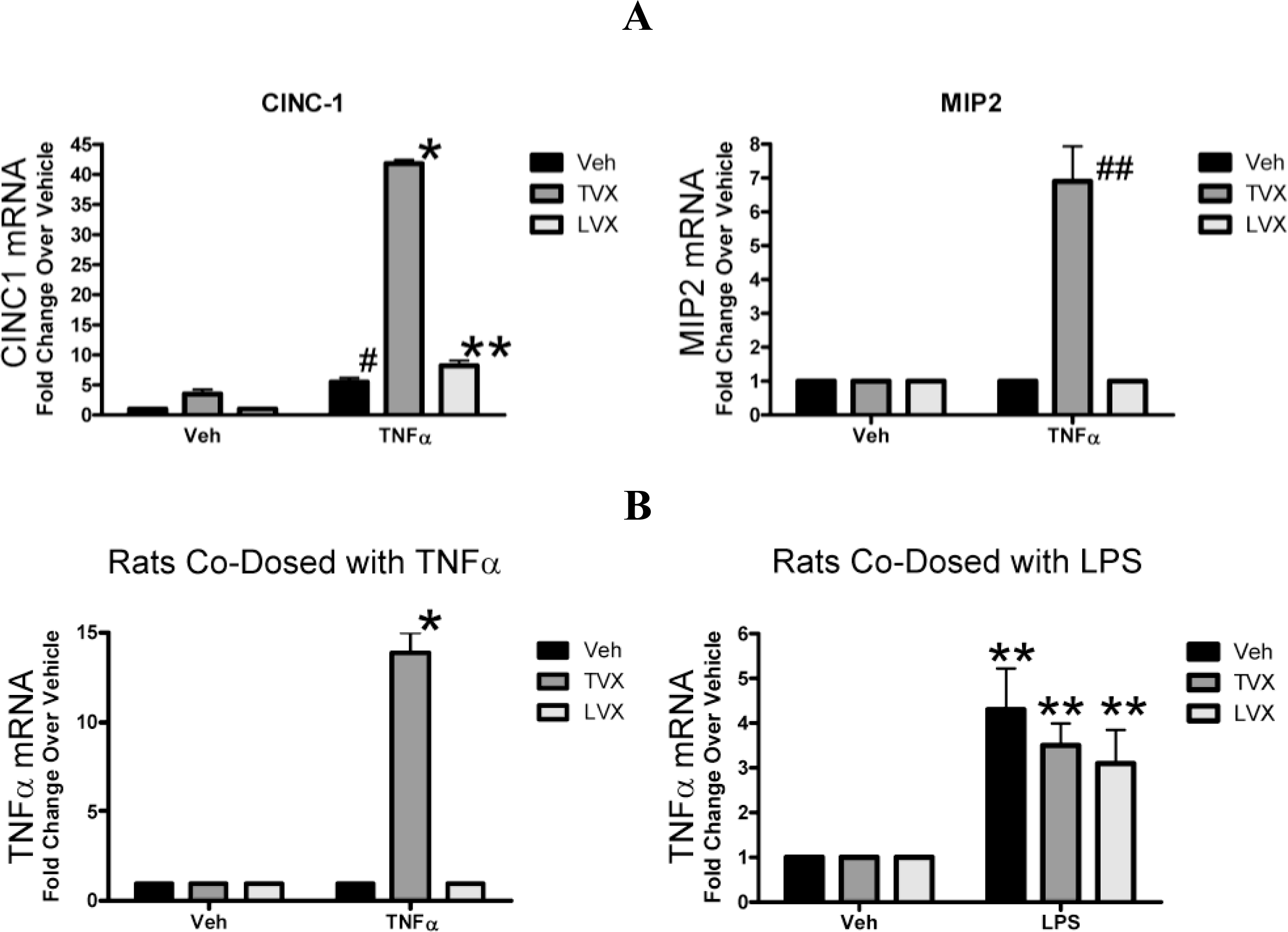
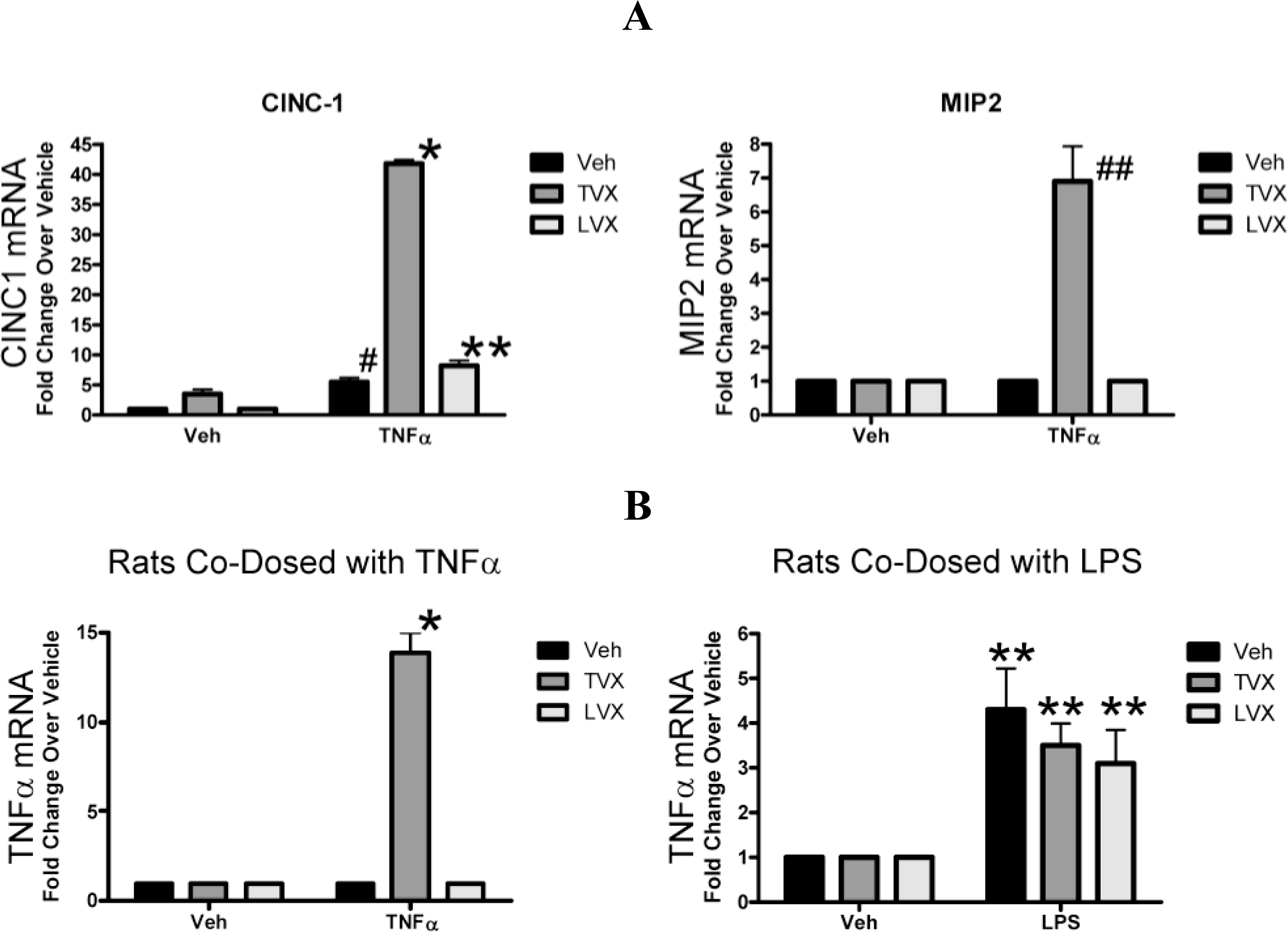
2.1.4. Evaluation of Fluoroquinolone Toxicity after TNFα Coadministration
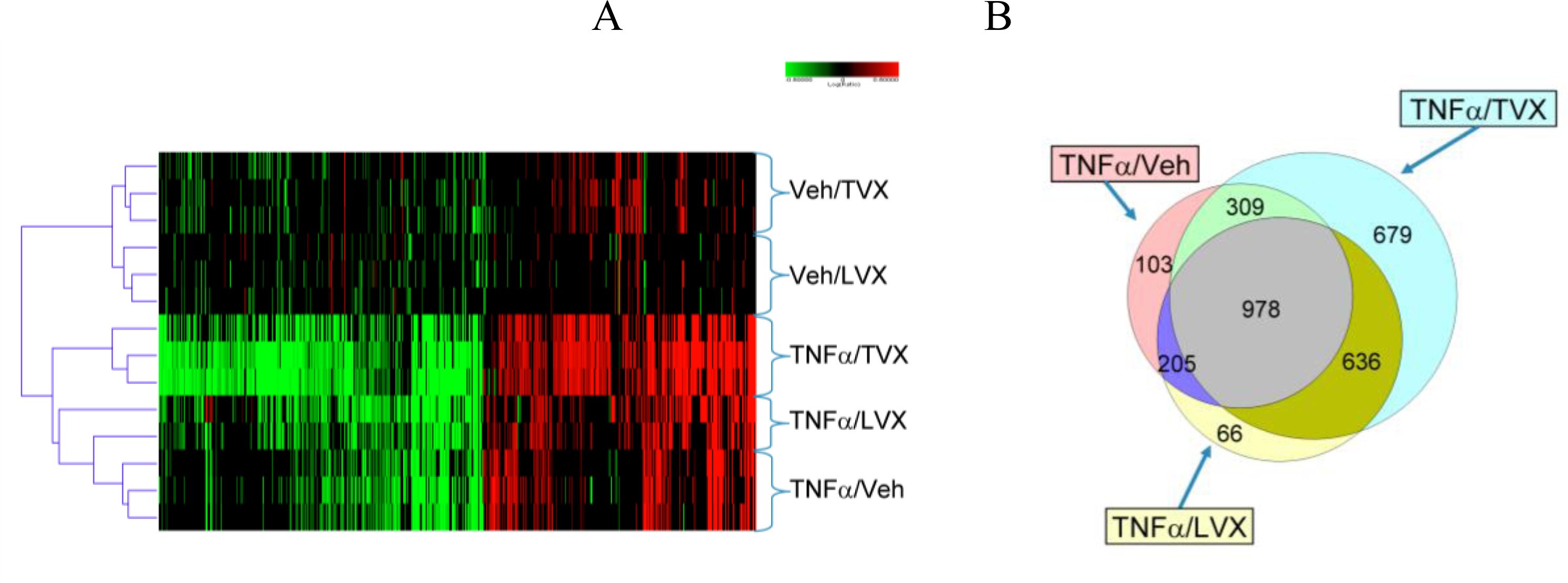
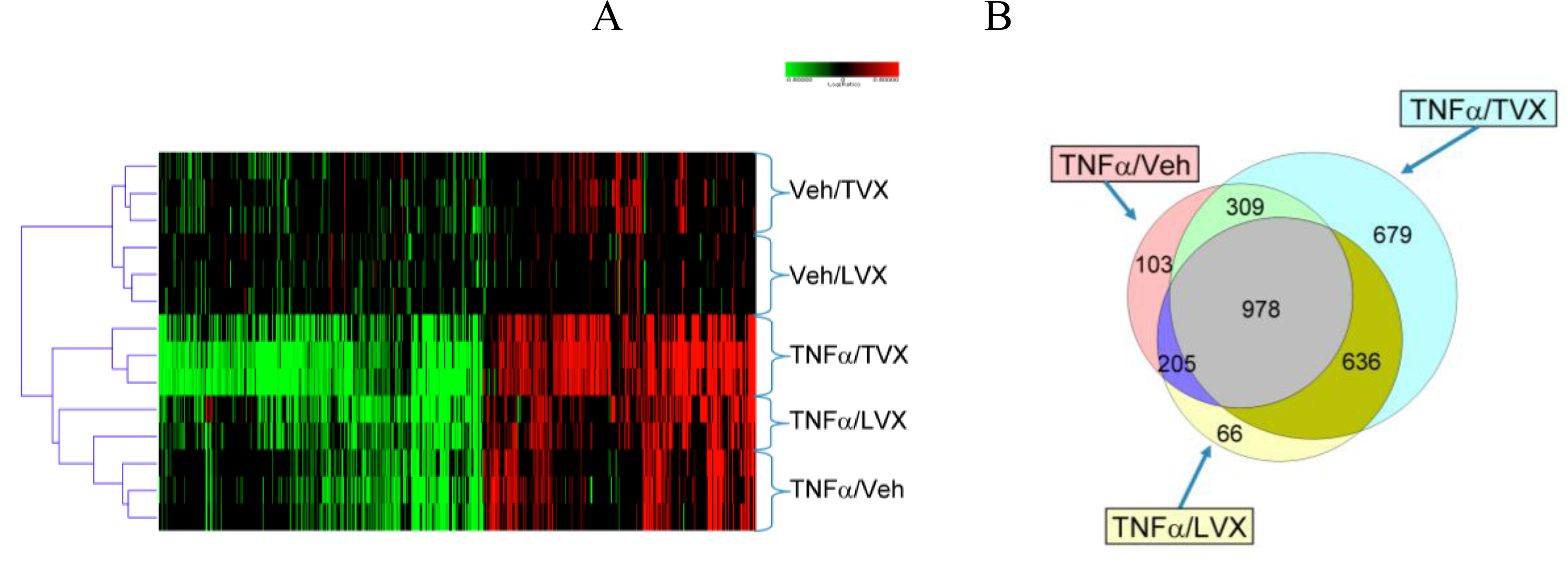
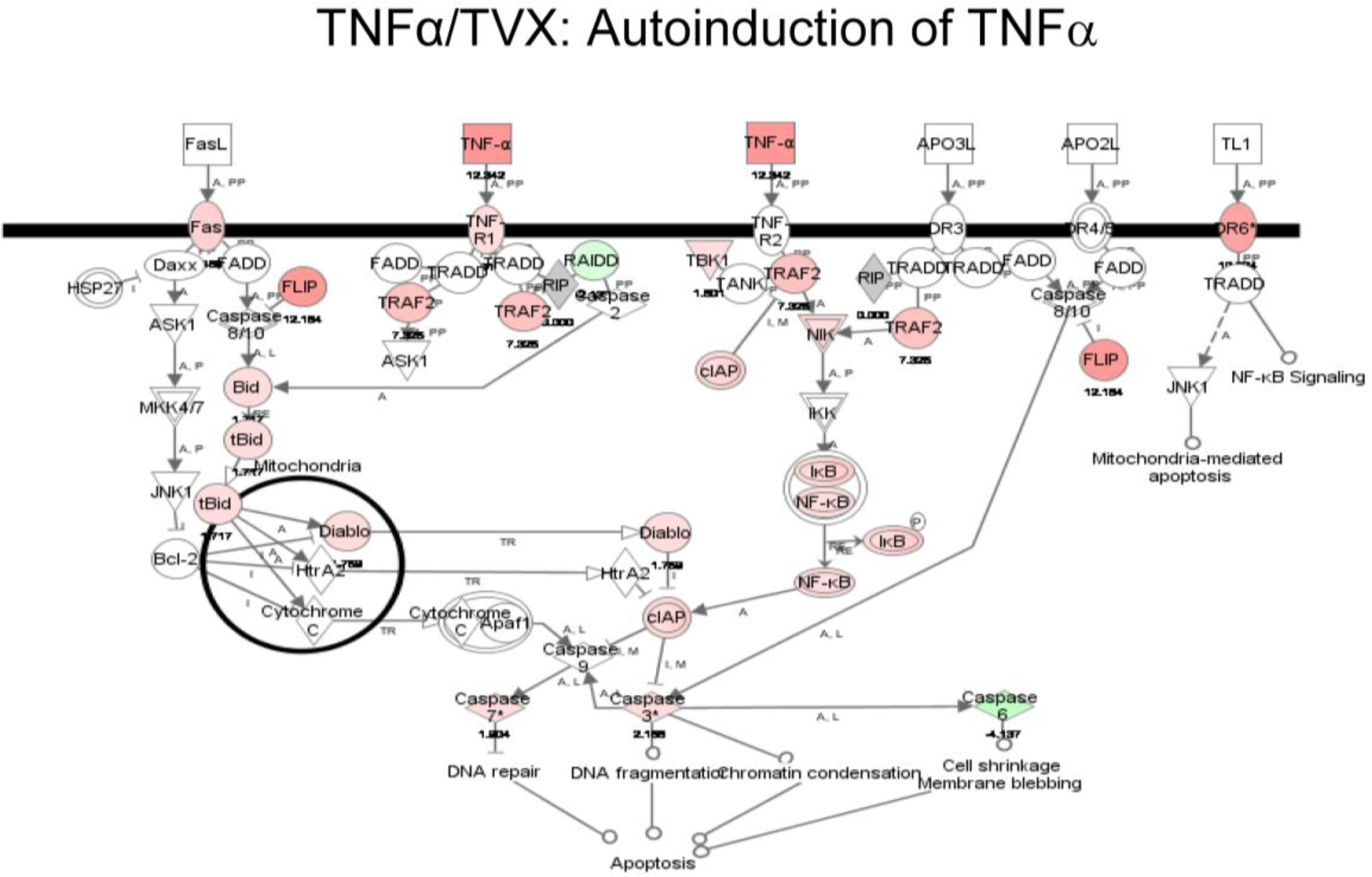
2.1.5. Comparison of Gene Expression Profiles Induced by TNFα/TVX and LPS/TVX
2.2. Discussion
3. Experimental Section
3.1. Administration of TNFα and Fluoroquinolone Drug
3.2. Administration of LPS
3.3. Serum Chemistry and Histopathology for the TNFα Study
3.4. RNA Preparation
3.5. Gene Array Analysis
3.6. Statistics and Pathway Analysis
4. Conclusions
Supplementary Data
ijms-11-04697-s001.xlsAcknowledgments
References
- Liguori, MJ; Waring, JF. Investigations toward enhanced understanding of hepatic idiosyncratic drug reactions. Exp. Opin. Drug Metab. Toxicol 2006, 2, 835–846. [Google Scholar]
- Roth, RA; Ganey, PE. Intrinsic versus idiosyncratic drug-induced hepatotoxicity--two villains or one? J. Pharmacol. Exp. Ther 2010, 332, 692–697. [Google Scholar]
- Deng, X; Stachlewitz, RF; Liguori, MJ; Blomme, EA; Waring, JF; Luyendyk, JP; Maddox, JF; Ganey, PE; Roth, RA. Modest inflammation enhances diclofenac hepatotoxicity in rats: Role of neutrophils and bacterial translocation. J. Pharmacol. Exp. Ther 2006, 319, 1191–1199. [Google Scholar]
- Waring, JF; Liguori, MJ; Luyendyk, JP; Maddox, JF; Ganey, PE; Stachlewitz, RF; North, C; Blomme, EA; Roth, RA. Microarray analysis of lipopolysaccharide potentiation of trovafloxacin-induced liver injury in rats suggests a role for proinflammatory chemokines and neutrophils. J. Pharmacol. Exp. Ther 2006, 316, 1080–1087. [Google Scholar]
- Luyendyk, JP; Maddox, JF; Cosma, GN; Ganey, PE; Cockerell, GL; Roth, RA. Ranitidine treatment during a modest inflammatory response precipitates idiosyncrasy-like liver injury in rats. J. Pharmacol. Exp. Ther 2003, 307, 9–16. [Google Scholar]
- Buchweitz, JP; Ganey, PE; Bursian, SJ; Roth, RA. Underlying endotoxemia augments toxic responses to chlorpromazine: Is there a relationship to drug idiosyncrasy? J. Pharmacol. Exp. Ther 2002, 300, 460–467. [Google Scholar]
- Zou, W; Beggs, KM; Sparkenbaugh, EM; Jones, AD; Younis, HS; Roth, RA; Ganey, PE. Sulindac metabolism and synergy with tumor necrosis factor-alpha in a drug-inflammation interaction model of idiosyncratic liver injury. J. Pharmacol. Exp. Ther 2009, 331, 114–121. [Google Scholar]
- Shaw, PJ; Hopfensperger, MJ; Ganey, PE; Roth, RA. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol. Sci 2007, 100, 259–266. [Google Scholar]
- Stahlmann, R. Clinical toxicological aspects of fluoroquinolones. Toxicol. Lett 2002, 127, 269–277. [Google Scholar]
- Blomme, EA; Yang, Y; Waring, JF. Use of toxicogenomics to understand mechanisms of drug-induced hepatotoxicity during drug discovery and development. Toxicol. Lett 2009, 186, 22–31. [Google Scholar]
- Shaw, PJ; Ganey, PE; Roth, RA. Trovafloxacin enhances the inflammatory response to a Gram-negative or a Gram-positive bacterial stimulus, resulting in neutrophil-dependent liver injury in mice. J. Pharmacol. Exp. Ther 2009, 330, 72–78. [Google Scholar]
- Shaw, PJ; Ditewig, AC; Waring, JF; Liguori, MJ; Blomme, EA; Ganey, PE; Roth, RA. Coexposure of mice to trovafloxacin and lipopolysaccharide, a model of idiosyncratic hepatotoxicity, results in a unique gene expression profile and interferon gamma-dependent liver injury. Toxicol. Sci 2009, 107, 270–280. [Google Scholar]
- De Castro, C; Molinaro, A; Lanzetta, R; Silipo, A; Parrilli, M. Lipopolysaccharide structures from Agrobacterium and Rhizobiaceae species. Carbohydr. Res 2008, 343, 1924–1933. [Google Scholar]
- Locksley, RM; Killeen, N; Lenardo, MJ. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar]
- Bradley, JR. TNF-mediated inflammatory disease. J. Pathol 2008, 214, 149–160. [Google Scholar]
- Deng, X; Luyendyk, JP; Ganey, PE; Roth, RA. Inflammatory stress and idiosyncratic hepatotoxicity: Hints from animal models. Pharmacol. Rev 2009, 61, 262–282. [Google Scholar]
- Shaw, PJ; Beggs, KM; Sparkenbaugh, EM; Dugan, CM; Ganey, PE; Roth, RA. Trovafloxacin enhances TNF-induced inflammatory stress and cell death signaling and reduces TNF clearance in a murine model of idiosyncratic hepatotoxicity. Toxicol. Sci 2009, 111, 288–301. [Google Scholar]
- Doyle, SL; O’Neill, LA. Toll-like receptors: From the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem. Pharmacol 2006, 72, 1102–1113. [Google Scholar]
- Mitchell, JA; Paul-Clark, MJ; Clarke, GW; McMaster, SK; Cartwright, N. Critical role of toll-like receptors and nucleotide oligomerisation domain in the regulation of health and disease. J. Endocrinol 2007, 193, 323–330. [Google Scholar]
- Rousseau, S; Papoutsopoulou, M; Symons, A; Cook, D; Lucocq, JM; Prescott, AR; O’Garra, A; Ley, SC; Cohen, P. TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNF alpha in LPS-stimulated macrophages. J. Cell Sci 2008, 121, 149–154. [Google Scholar]
- Janssens, S; Beyaert, R. Role of Toll-like receptors in pathogen recognition. Clin. Microbiol. Rev 2003, 16, 637–646. [Google Scholar]
- Kol, A; Bourcier, T; Lichtman, AH; Libby, P. Chlamydial and human heat shock protein 60s activate human vascular endothelium, smooth muscle cells, and macrophages. J. Clin. Invest 1999, 103, 571–577. [Google Scholar]
- Pulendran, B. Modulating vaccine responses with dendritic cells and Toll-like receptors. Immunol. Rev 2004, 199, 227–250. [Google Scholar]
- Loegering, DJ; Drake, JR; Banas, JA; McNealy, TL; Mc Arthur, DG; Webster, LM; Lennartz, MR. Francisella tularensis LVS grown in macrophages has reduced ability to stimulate the secretion of inflammatory cytokines by macrophages in vitro. Microb. Pathog 2006, 41, 218–225. [Google Scholar]
- Ribot, WJ; Panchal, RG; Brittingham, KC; Ruthel, G; Kenny, TA; Lane, D; Curry, B; Hoover, TA; Friedlander, AM; Bavari, S. Anthrax lethal toxin impairs innate immune functions of alveolar macrophages and facilitates Bacillus anthracis survival. Infect. Immun 2006, 74, 5029–5034. [Google Scholar]
- Muriel, P. NF-kappaB in liver diseases: A target for drug therapy. J. Appl. Toxicol 2009, 29, 91–100. [Google Scholar]
- Xia, ZP; Chen, ZJ. TRAF2: A double-edged sword. Sci STKE 2005, 2005, pe7. [Google Scholar]
- Shaw, PJ; Ganey, PE; Roth, RA. Tumor necrosis factor alpha is a proximal mediator of synergistic hepatotoxicity from trovafloxacin/lipopolysaccharide coexposure. J. Pharmacol. Exp. Ther 2009, 328, 62–68. [Google Scholar]
- Adams, DH; Ju, C; Ramaiah, SK; Uetrecht, J; Jaeschke, H. Mechanisms of immune-mediated liver injury. Toxicol. Sci 2010, 115, 307–321. [Google Scholar]
- Spriggs, D; Imamura, K; Rodriguez, C; Horiguchi, J; Kufe, DW. Induction of tumor necrosis factor expression and resistance in a human breast tumor cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 6563–6566. [Google Scholar]
- Bader, T; Nettesheim, P. Tumor necrosis factor-alpha modulates the expression of its p60 receptor and several cytokines in rat tracheal epithelial cells. J. Immunol 1996, 157, 3089–3096. [Google Scholar]
- Spriggs, DR; Sherman, ML; Imamura, K; Mohri, M; Rodriguez, C; Robbins, G; Kufe, DW. Phospholipase A2 activation and autoinduction of tumor necrosis factor gene expression by tumor necrosis factor. Cancer Res 1990, 50, 7101–7107. [Google Scholar]
- Ganter, B; Snyder, RD; Halbert, DN; Lee, MD. Toxicogenomics in drug discovery and development: Mechanistic analysis of compound/class-dependent effects using the DrugMatrix database. Pharmacogenomics 2006, 7, 1025–1044. [Google Scholar]
- Luyendyk, JP; Lehman-McKeeman, LD; Nelson, DM; Bhaskaran, VM; Reilly, TP; Car, BD; Cantor, GH; Maddox, JF; Ganey, PE; Roth, RA. Unique gene expression and hepatocellular injury in the lipopolysaccharide-ranitidine drug idiosyncrasy rat model: Comparison with famotidine. Toxicol. Sci 2006, 90, 569–585. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Both TNFα/TVX & LPS/TVX | ||||
|---|---|---|---|---|
| Pathway Name | TNF/TVX % Impact | TNF/TVX P-Value | LPS/TVX % Impact | LPS/TVX P-Value |
| Tight Junction Signaling | 26.1% | 0.001 | 18.5% | 0.021 |
| MIF Regulation of Innate Immunity | 33.3% | 0.003 | 22.2% | 0.039 |
| Dendritic Cell Maturation | 25.3% | 0.004 | 20.0% | 0.013 |
| NFAT Regulation of Immune Response | 24.4% | 0.011 | 17.9% | 0.058 |
| fMLP Signaling in Neutrophils | 26.2% | 0.013 | 19.7% | 0.051 |
| IL-12 Signaling in Macrophages | 23.9% | 0.018 | 20.9% | 0.014 |
| Glucocorticoid Receptor Signaling | 20.5% | 0.021 | 17.9% | 0.012 |
| p53 Signaling | 22.0% | 0.025 | 22.0% | 0.004 |
| B Cell Receptor Signaling | 20.5% | 0.040 | 18.2% | 0.023 |
| Role of Macrophages, Fibroblasts and Endothelial Cells in RA | 18.3% | 0.040 | 17.6% | 0.005 |
| Molecular Mechanisms of Cancer | 17.8% | 0.044 | 14.9% | 0.043 |
| TNFα/TVX | ||
|---|---|---|
| Pathway Name | % Impact | P-Value |
| ATM Signaling | 34.4% | 0.001 |
| Hypoxia Signaling in the Cardiovascular System | 32.0% | 0.001 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 37.0% | 0.002 |
| CD28 Signaling in T Helper Cells | 27.9% | 0.003 |
| HMGB1 Signaling | 27.0% | 0.004 |
| Role of PKR in Interferon Induction and Antiviral Response | 32.3% | 0.006 |
| Androgen Signaling | 26.1% | 0.007 |
| Activation of IRF by Pattern Recognition Receptors | 27.8% | 0.008 |
| NF-κB Signaling | 23.8% | 0.009 |
| IL-10 Signaling | 27.5% | 0.010 |
| CD27 Signaling in Lymphocytes | 27.3% | 0.010 |
| p38 MAPK Signaling | 24.2% | 0.012 |
| 4-1BB Signaling in T Lymphocytes | 31.8% | 0.013 |
| Death Receptor Signaling | 25.0% | 0.017 |
| Cholecystokinin/Gastrin-mediated Signaling | 24.2% | 0.024 |
| iCOS-iCOSL Signaling in T Helper Cells | 24.5% | 0.025 |
| Relaxin Signaling | 23.5% | 0.025 |
| Role of BRCA1 in DNA Damage Response | 25.0% | 0.026 |
| Role of RIG1-like Receptors in Antiviral Innate Immunity | 27.3% | 0.026 |
| Production of Nitric Oxide and ROS in Macrophages | 21.5% | 0.028 |
| ILK Signaling | 21.1% | 0.031 |
| Cdc42 Signaling | 21.4% | 0.040 |
| Angiopoietin Signaling | 25.0% | 0.042 |
| AMPK Signaling | 23.8% | 0.045 |
| Pattern Recognition Receptors in Bacteria/Virus | 20.5% | 0.046 |
| Phospholipase C Signaling | 17.9% | 0.049 |
| Hepatic Cholestasis | 19.8% | 0.050 |
| Protein Kinase A Signaling | 18.5% | 0.060 |
| B Cell Activating Factor Signaling | 24.0% | 0.062 |
| LPS/TVX | ||
|---|---|---|
| Pathway Name | % Impact | P-Value |
| Chronic Myeloid Leukemia Signaling | 23.1% | 0.006 |
| LPS-stimulated MAPK Signaling | 25.5% | 0.006 |
| PDGF Signaling | 23.4% | 0.017 |
| Communication between Innate and Adaptive Immune Cells | 25.0% | 0.018 |
| PPAR Signaling | 21.9% | 0.019 |
| VDR/RXR Activation | 23.1% | 0.026 |
| Cytokines in Mediating Communication between Immune Cells | 31.2% | 0.028 |
| Toll-like Receptor Signaling | 20.0% | 0.031 |
| GM-CSF Signaling | 24.4% | 0.032 |
| Cleavage and Polyadenylation of Pre-mRNA | 37.5% | 0.034 |
| Fc Epsilon RI Signaling | 19.6% | 0.039 |
| Cell Cycle: G1/S Checkpoint Regulation | 20.5% | 0.044 |
| Estrogen Receptor Signaling | 18.2% | 0.046 |
| T Cell Receptor Signaling | 18.9% | 0.049 |
| CD40 Signaling | 21.1% | 0.051 |
| Wnt/β-catenin Signaling | 17.4% | 0.051 |
| PI3K/AKT Signaling | 18.3% | 0.056 |
| TREM1 Signaling | 21.9% | 0.056 |
| Regulation of eIF4 and p70S6K Signaling | 19.1% | 0.058 |
| Mitochondrial Dysfunction | 17.6% | 0.062 |
| Cell Cycle Regulation by BTG Family Proteins | 25.0% | 0.063 |
Non-Standard Abbreviations:
| IDR | Idiosyncratic drug reaction |
| LPS | lipopolysaccharide |
| TNFα | tumor necrosis factor-α |
| TVX | trovafloxacin |
| LVX | levofloxacin |
| OPS | O-specific polysaccharide |
| ALT | alanine amino transferase |
| AST | aspartate amino transferase |
| GGT | gamma glutamyltransferase |
| ALP | alkaline phosphatase |
| TBIL | total bilirubin |
| PCA | principal component analysis |
| interferon | IFN |
| PMN | neutrophil |
| ER | endoplasmic reticulum |
| ROS | reactive oxygen species |
| MAP3Ks | mitogen-activated protein kinase kinase kinase |
| IL-12 | interleukin-12 |
| ATM | ataxia telangiectasia-mutated |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liguori, M.J.; Ditewig, A.C.; Maddox, J.F.; Luyendyk, J.P.; Lehman-McKeeman, L.D.; Nelson, D.M.; Bhaskaran, V.M.; Waring, J.F.; Ganey, P.E.; Roth, R.A.; et al. Comparison of TNFα to Lipopolysaccharide as an Inflammagen to Characterize the Idiosyncratic Hepatotoxicity Potential of Drugs: Trovafloxacin as an Example. Int. J. Mol. Sci. 2010, 11, 4697-4714. https://doi.org/10.3390/ijms11114697
Liguori MJ, Ditewig AC, Maddox JF, Luyendyk JP, Lehman-McKeeman LD, Nelson DM, Bhaskaran VM, Waring JF, Ganey PE, Roth RA, et al. Comparison of TNFα to Lipopolysaccharide as an Inflammagen to Characterize the Idiosyncratic Hepatotoxicity Potential of Drugs: Trovafloxacin as an Example. International Journal of Molecular Sciences. 2010; 11(11):4697-4714. https://doi.org/10.3390/ijms11114697
Chicago/Turabian StyleLiguori, Michael J., Amy C. Ditewig, Jane F. Maddox, James P. Luyendyk, Lois D. Lehman-McKeeman, David M. Nelson, Vasanthi M. Bhaskaran, Jeffrey F. Waring, Patricia E. Ganey, Robert A. Roth, and et al. 2010. "Comparison of TNFα to Lipopolysaccharide as an Inflammagen to Characterize the Idiosyncratic Hepatotoxicity Potential of Drugs: Trovafloxacin as an Example" International Journal of Molecular Sciences 11, no. 11: 4697-4714. https://doi.org/10.3390/ijms11114697