3,4-Methylenedioxymethamphetamine Alters Left Ventricular Function and Activates Nuclear Factor-Kappa B (NF-kB) in a Time and Dose Dependent Manner

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. In Vivo Experiments
2.2. In Vitro Experiments
2.2.1. Reactive Oxygen Species Assay
2.2.2. ELISA for NF-κB
3. Results
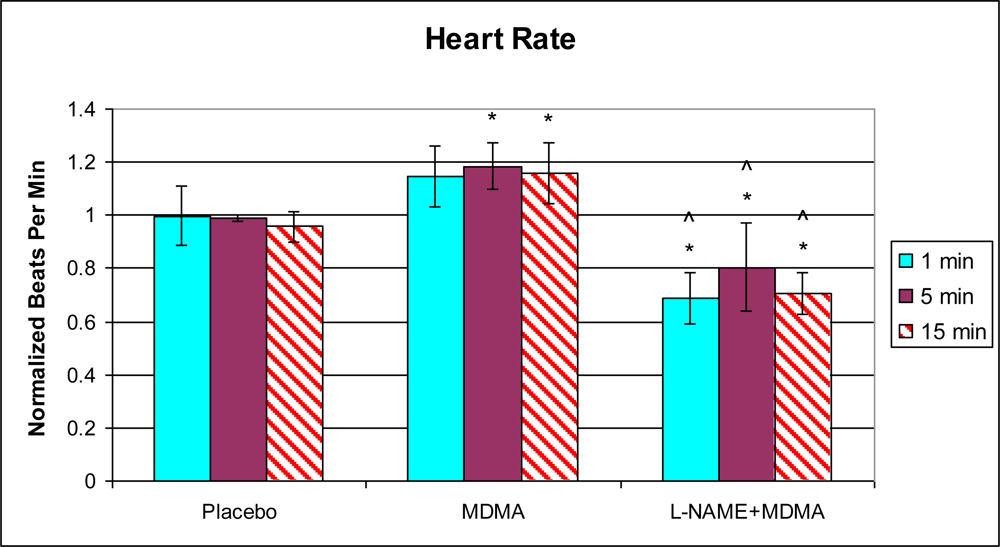
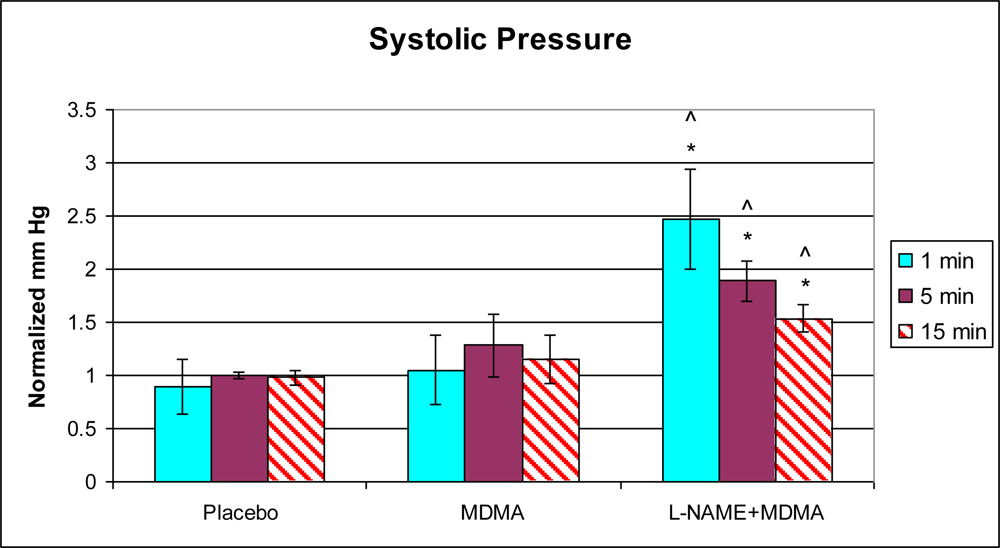
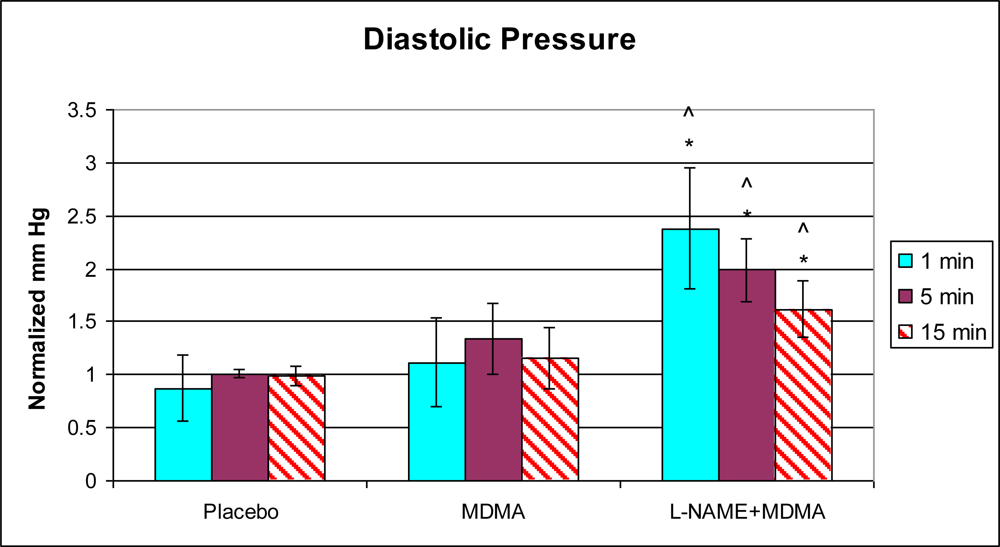
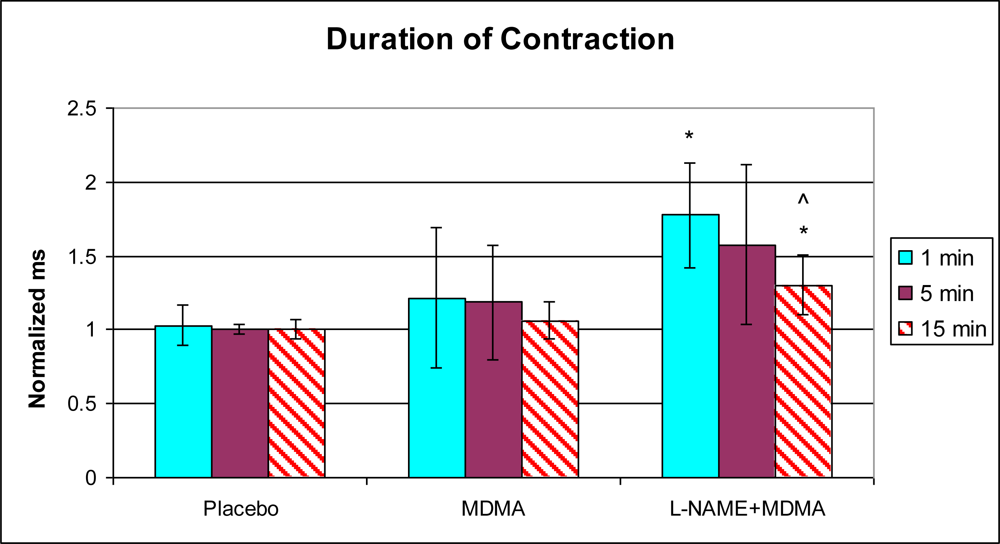
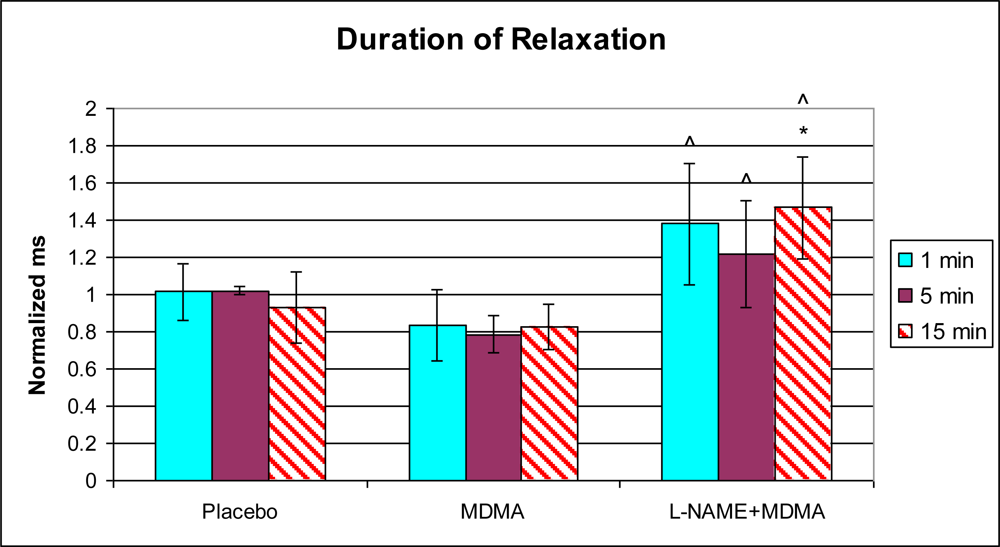
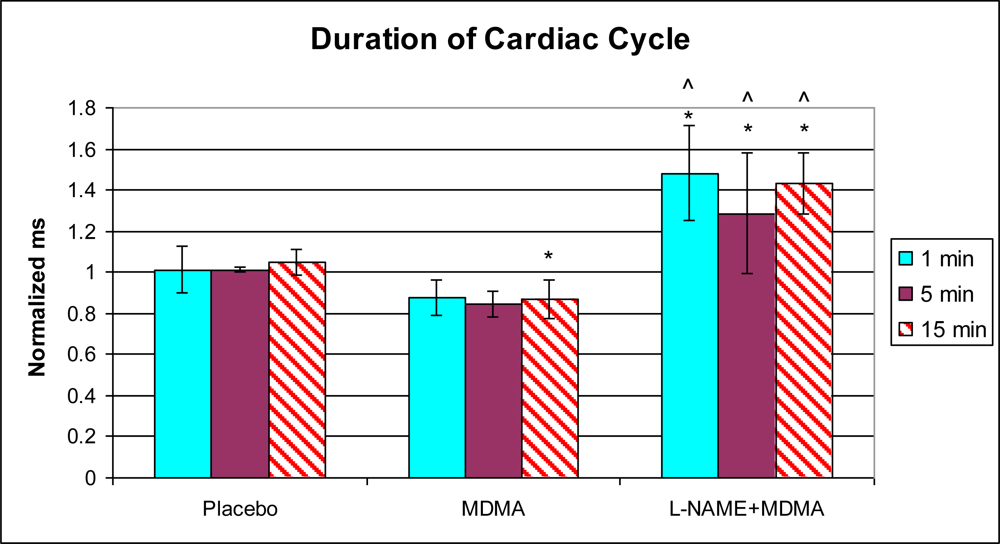
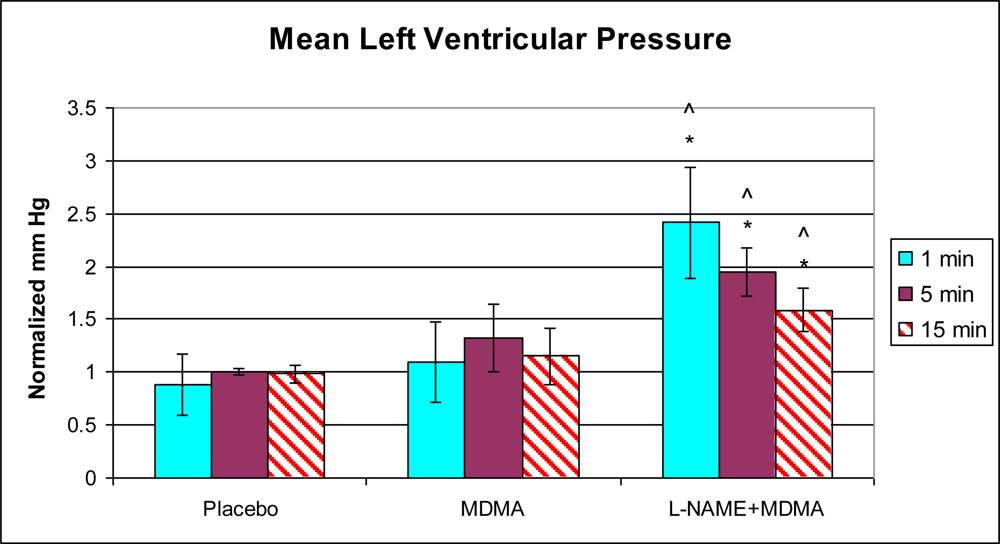
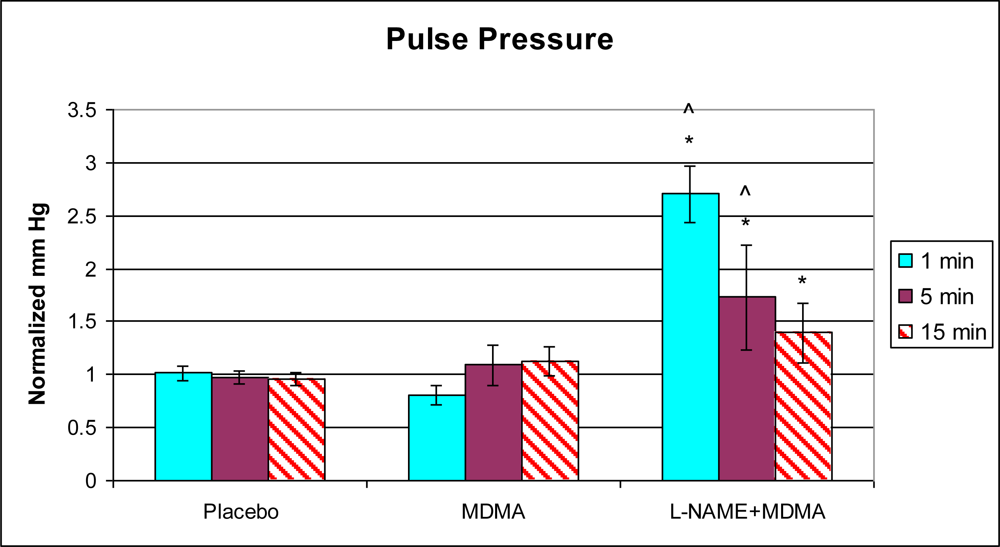
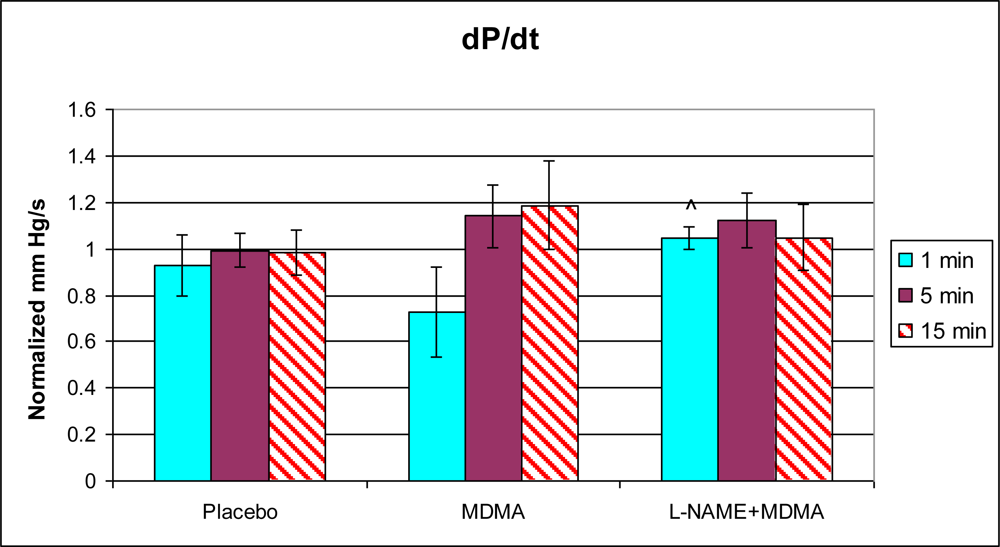
3.1. In Vivo Experiments
3.2. In Vitro Experiments
3.2.1. Reactive Oxygen Species Assay
3.2.2. ELISA for NF-κB
4. Discussion
4.1. Effects of MDMA
4.2. MDMA and L-NAME
5. Conclusions
Acknowledgments
References
- Kalant, H. The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. Can. Med. Assoc. J 2001, 165, 917–928. [Google Scholar]
- Lester, SJ; Baggott, M; Welm, S; Schiller, NB; Jones, RT; Foster, E; Mendelson, J. Cardiovascular effects of 3,4-methylenediosymethamphetamine. A double-blind, placebo-controlled trial. Ann. Intern. Med 2000, 133, 969–973. [Google Scholar]
- Badon, LA; Hicks, A; Lord, K; Ogden, BA; Meleg-Smith, S; Varner, KJ. Changes in cardiovascular responsiveness and cardiotoxicity elicited during binge administration of ecstasy. J. Pharmacol. Exp. Ther 2002, 302, 898–907. [Google Scholar]
- Brody, S; Krause, C; Veit, R; Rau, H. Cardiovascular autonomic dysregulation in users of MDMA (“ecstasy”). Psychopharmacology 1998, 136, 390–393. [Google Scholar]
- Vandeputte, C; Docherty, JR. Vascular actions of 3,4-methylenedioxymethamphetamine in alpha(2A/D)-adrenoceptor knockout mice. Eur. J. Pharmacol 2002, 457, 45–49. [Google Scholar]
- Carvalho, M; Remiao, F; Milhazes, N; Borges, F; Fernandes, E; Monteiro; Mdo, C; Goncalves, MJ; Seabra, V; Amado, F; Carvalho, F; Bastos, ML. Metabolism is required for the expression of ecstasy-induced cardiotoxicity in vitro. Chem. Res. Toxicol 2004, 17, 623–632. [Google Scholar]
- Sheridan, RD; Turner, SR; Cooper, GJ; Tattersall, JE. Effects of seven drugs of abuse on action potential repolarisation in sheep cardiac Purkinje fibres. Eur. J. Pharmacol 2005, 511, 99–107. [Google Scholar]
- Patel, MM; Belson, MG; Wright, D; Lu, H; Heninger, M; Miller, MA. Methylenedioxymethamphetamine (ecstasy)-related myocardial hypertrophy: An autopsy study. Resuscitation 2005, 66, 197–202. [Google Scholar]
- Bexis, S; Docherty, JR. Effects of MDMA, MDA and MDEA on blood pressure, heart rate, locomotor activity and body temperature in the rat involve alpha-adrenoceptors. Br. J. Pharmacol 2006, 147, 926–934. [Google Scholar]
- McNamara, R; Maginn, M; Harkin, A. Caffeine induces a profound and persistent tachycardia in response to MDMA (“Ecstasy”) administration. Eur. J. Pharmacol 2007, 555, 194–198. [Google Scholar]
- Tiangco, DA; Lattanzio, FA, Jr; Osgood, CJ; Beebe, SJ; Kerry, JA; Hargrave, BY. 3,4-Methylenedioxymethamphetamine activates nuclear factor-kappaB, increases intracellular calcium, and modulates gene transcription in rat heart cells. Cardiovasc. Toxicol 2005, 5, 301–310. [Google Scholar]
- Muth, JN; Bodi, I; Lewis, W; Varadi, G; Schwartz, A. A Ca(2+)-dependent transgenic model of cardiac hypertrophy: A role for protein kinase Calpha. Circulation 2001, 103, 140–147. [Google Scholar]
- Sen, R; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar]
- Kaltschmidt, C; Kaltschmidt, B; Neumann, H; Wekerle, H; Baeuerle, PA. Constitutive NF-kappa B activity in neurons. Mol. Cell. Biol 1994, 14, 3981–3992. [Google Scholar]
- Lawrence, R; Chang, LJ; Siebenlist, U; Bressler, P; Sonenshein, GE. Vascular smooth muscle cells express a constitutive NF-kappa B-like activity. J. Biol. Chem 1994, 269, 28913–28918. [Google Scholar]
- Grimm, S; Baeuerle, PA. The inducible transcription factor NF-κB: Structure-function relationship of its protein subunits. Biochem. J 1993, 290, 297–308. [Google Scholar]
- Baeuerle, PA; Baltimore, D. I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science 1988, 242, 540–546. [Google Scholar]
- Baeuerle, PA; Baltimore, D. Activation of DNA-binding activity in an apparently cytoplasmic precursor of the NF-κB transcription factor. Cell 1988, 53, 211–217. [Google Scholar]
- Henkel, T; Machleidt, T; Alkalay, I; Kronke, M; Ben-Neriah, Y; Baeuerle, PA. Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature 1993, 365, 182–185. [Google Scholar]
- Palombella, VJ; Rando, OJ; Goldberg, AL; Maniatis, T. The ubiquitin-proteasome pathway is required for processing the NF-kappa B1 precursor protein and the activation of NF-kappa B. Cell 1994, 78, 773–785. [Google Scholar]
- Traenckner, EB; Pahl, HL; Henkel, T; Schmidt, KN; Wilk, S; Baeuerle, PA. Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha proteolysis and NF-kappa B activation in response to diverse stimuli. J. EMBO 1995, 14, 2876–2883. [Google Scholar]
- Thompson, JE; Phillips, RJ; Erdjument-Bromage, H; Tempst, P; Ghosh, S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell 1995, 80, 573–582. [Google Scholar]
- Grilli, M; Chiu, JJ; Lenardo, MJ. NF-kappa B and Rel: Participants in a multiform transcriptional regulatory system. Int. Rev. Cytol 1993, 143, 1–62. [Google Scholar]
- Siebenlist, U; Franzoso, G; Brown, K. Structure, regulation and function of NF-kappa B. Annu. Rev. Cell. Biol 1994, 10, 405–455. [Google Scholar]
- Baeuerle, PA; Baltimore, D. NF-kappa B: Ten years after. Cell 1996, 87, 13–20. [Google Scholar]
- Landry, DB; Couper, LL; Bryant, SR; Lindner, V. Activation of the NF-kappa B and I kappa B system in smooth muscle cells after rat arterial injury. Induction of vascular cell adhesion molecule-1 and monocyte chemoattractant protein-1. Am. J. Pathol 1997, 151, 1085–1095. [Google Scholar]
- Brach, MA; Hass, R; Sherman, ML; Gunji, H; Weichselbaum, R; Kufe, D. Ionizing radiation induces expression and binding activity of the nuclear factor kappa B. J. Clin. Invest 1991, 88, 691–695. [Google Scholar]
- Schreck, R; Rieber, P; Baeuerle, PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. J. EMBO 1991, 10, 2247–2258. [Google Scholar]
- Hargrave, BY; Tiangco, DA; Lattanzio, FA; Beebe, SJ. Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-κB in transiently cotransfected heart cells. Cardiovas. Toxicol 2003, 3, 141–151. [Google Scholar]
- Peng, M; Huang, L; Xie, ZJ; Huang, WH; Askari, A. Oxidant-induced activations of nuclear factor-kappa B and activator protein-1 in cardiac myocytes. Cell. Mol. Biol. Res 1995, 41, 189–197. [Google Scholar]
- Hattori, Y; Akimoto, K; Murakami, Y; Kasai, K. Pyrrolidine dithiocarbamate inhibits cytokine-induced VCAM-1 gene expression in rat cardiac myocytes. Mol. Cell. Biochem 1997, 177, 177–181. [Google Scholar]
- Kacimi, R; Karliner, JS; Koudssi, F; Long, CS. Expression and regulation of adhesion molecules in cardiac cells by cytokines: Response to acute hypoxia. Circ. Res 1998, 82, 576–586. [Google Scholar]
- Mustapha, S; Kirshner, A; De Moissac, D; Kirshenbaum, LA. A direct requirement of nuclear factor-kappa B for suppression of apoptosis in ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol 2000, 279, H939–H945. [Google Scholar]
- Montiel-Duarte, C; Ansorena, E; Lopez-Zabalza, MJ; Cenarruzabeitia, E; Iraburu, MJ. Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxymethamphetamine (“ecstasy”) on hepatic stellate cells. Biochem. Pharmacol 2004, 67, 1025–1033. [Google Scholar]
- Shankaran, M; Yamamoto, BK; Gudelsky, GA. Involvement of the serotonin transporter in the formation of hydroxyl radicals induced by 3,4-methylenedioxymethamphetamine. Eur. J. Pharmacol 1999, 385, 103–110. [Google Scholar]
- Shenouda, SK; Lord, KC; McIlwain, E; Lucchesi, PA; Varner, KJ. Ecstasy produces left ventricular dysfunction and oxidative stress in rats. Cardiovasc. Res 2008, 79, 662–670. [Google Scholar]
- Quaini, F; Cigola, E; Lagrasta, C; Saccani, G; Quaini, E; Rossi, C; Olivetti, G; Anversa, P. End-stage cardiac failure in humans is coupled with the induction of proliferating cell nuclear antigen and nuclear mitotic division in ventricular myocytes. Circ. Res 1994, 75, 1050–1063. [Google Scholar]
- Wong, SC; Fukuchi, M; Melnyk, P; Rodger, I; Giaid, A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation 1998, 98, 100–103. [Google Scholar]
- Smith, ML; Chen, IT; Zhan, Q; Bae, I; Chen, CY; Gilmer, TM; Kastan, MB; O’Connor, PM; Fornace, AJ, Jr. Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 1994, 266, 1376–1380. [Google Scholar]
- Ng, JM; Vermeulen, W; van der Horst, GT; Bergink, S; Sugasawa, K; Vrieling, H; Hoeijmakers, JH. A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev 2003, 17, 1630–1645. [Google Scholar]
- Kinugawa, K; Shimizu, T; Yao, A; Kohmoto, O; Serizawa, T; Takahashi, T. Transcriptional regulation of inducible nitric oxide synthase in cultured neonatal rat cardiac myocytes. Circ. Res 1997, 81, 911–921. [Google Scholar]
- Liang, YJ; Lai, LP; Wang, BW; Juang, SJ; Chang, CM; Leu, JG; Shyu, KG. Mechanical stress enhances serotonin 2B receptor modulating brain natriuretic peptide through nuclear factor-kappaB in cardiomyocytes. Cardiovasc. Res 2006, 72, 303–312. [Google Scholar]
- Palmer, RM; Ferrige, AG; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar]
- Rees, DD; Palmer, RM; Moncada, S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar]
- Mitchell, JA; Förstermann, U; Warner, TD; Pollock, JS; Schmidt, HH; Heller, M; Murad, F. Endothelial cells have a particulate enzyme system responsible for EDRF formation: Measurement by vascular relaxation. Biochem. Biophys. Res. Commun 1991, 176, 1417–1423. [Google Scholar]
- Rekhter, MD; Zhang, K; Narayanan, AS; Phan, S; Schork, MA; Gordon, D. Type I collagen gene expression in human atherosclerosis. Localization to specific plaque regions. Am. J. Pathol 1993, 143, 1634–1648. [Google Scholar]
- González-Santiago, L; López-Ongil, S; Rodríguez-Puyol, M; Rodríguez-Puyol, D. Decreased nitric oxide synthesis in human endothelial cells cultured on type I collagen. Circ. Res 2002, 90, 539–545. [Google Scholar]
- Samyn, N; De Boeck, G; Wood, M; Lamers, CT; De Waard, D; Brookhuis, KA; Verstraete, AG; Riedel, WJ. Plasma, oral fluid and sweat wipe ecstasy concentrations in controlled and real life conditions. Forensic Sci. Int 2002, 128, 90–97. [Google Scholar]
- de la Torre, R; Farré, M; Roset, PN; Lopez, CH; Mas, M; Ortuño, J; Menoyo, E; Pizarro, N; Segura, J; Cami, J. Pharmacology of MDMA in humans. Ann. N. Y. Acad. Sci 2000, 914, 225–237. [Google Scholar]
- Maurer, HH; Bickeboeller-Friedrich, J; Kraemer, T; Peters, FT. Toxicokinetics and analytical toxicology of amphetamine-derived designer drugs (‘Ecstasy’). Toxicol Lett 2000, 112–113, 133–142. [Google Scholar]
- de la Torre, R; Farré, M; Ortuño, J; Mas, M; Brenneisen, R; Roset, PN; Segura, J; Camí, J. Non-linear pharmacokinetics of MDMA (‘ecstasy’) in humans. Br. J. Clin. Pharmacol 2000, 49, 104–109. [Google Scholar]
- Capela, JP; Carmo, H; Remião, F; Bastos, ML; Meisel, A; Carvalho, F. Molecular and cellular mechanisms of ecstasy-induced neurotoxicity: An overview. Mol. Neurobiol 2009, 39, 210–271. [Google Scholar]
- Griendling, KK; Sorescu, D; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res 2000, 86, 494–501. [Google Scholar]
- Dhalla, NS; Afzal, N; Beamish, RE; Naimark, B; Takeda, N; Nagano, M. Pathophysiology of cardiac dysfunction in congestive heart failure. Can. J. Cardiol 1993, 9, 873–887. [Google Scholar]
- Shenouda, SK; Carvalho, F; Varner, KJ. The cardiovascular and cardiac actions of ecstasy and its metabolites. Curr. Pharm. Biotechnol 2010, 11, 470–475. [Google Scholar]
- Dawn, B; Bolli, R. Role of nitric oxide in myocardial preconditioning. Ann. N. Y. Acad. Sci 2002, 962, 18–41. [Google Scholar]
- Shindo, T; Ikeda, U; Ohkawa, F; Kawahara, Y; Yokoyama, M; Shimada, K. Nitric oxide synthesis in cardiac myocytes and fibroblasts by inflammatory cytokines. Cardiovasc. Res 1995, 29, 813–819. [Google Scholar]
- Rees, DD; Palmer, RM; Schulz, R; Hodson, HF; Moncada, S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br. J. Pharmacol 1990, 101, 746–752. [Google Scholar]
- Gozal, D; Torres, JE; Gozal, YM; Littwin, SM. Effect of nitric oxide synthase inhibition on cardiorespiratory responses in the conscious rat. J. Appl. Physiol 1996, 81, 2068–2077. [Google Scholar]
- Gardiner, SM; Compton, AM; Kemp, PA; Bennett, T. Regional and cardiac haemodynamic effects of NG-nitro-L-arginine methyl ester in conscious, Long Evans rats. Br. J. Pharmacol 1990, 101, 625–631. [Google Scholar]
- Shesely, EG; Maeda, N; Kim, HS; Desai, KM; Krege, JH; Laubach, VE; Sherman, PA; Sessa, WC; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar]
- Liu, JL; Murakami, H; Zucker, IH. Effects of NO on baroreflex control of heart rate and renal nerve activity in conscious rabbits. Am. J. Physiol 1996, 270, R1361–R1370. [Google Scholar]













© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tiangco, D.A.; Halcomb, S.; Lattanzio, F.A., Jr.; Hargrave, B.Y. 3,4-Methylenedioxymethamphetamine Alters Left Ventricular Function and Activates Nuclear Factor-Kappa B (NF-kB) in a Time and Dose Dependent Manner. Int. J. Mol. Sci. 2010, 11, 4843-4863. https://doi.org/10.3390/ijms11124743
Tiangco DA, Halcomb S, Lattanzio FA Jr., Hargrave BY. 3,4-Methylenedioxymethamphetamine Alters Left Ventricular Function and Activates Nuclear Factor-Kappa B (NF-kB) in a Time and Dose Dependent Manner. International Journal of Molecular Sciences. 2010; 11(12):4843-4863. https://doi.org/10.3390/ijms11124743
Chicago/Turabian StyleTiangco, David A., Sapna Halcomb, Frank A. Lattanzio, Jr., and Barbara Y. Hargrave. 2010. "3,4-Methylenedioxymethamphetamine Alters Left Ventricular Function and Activates Nuclear Factor-Kappa B (NF-kB) in a Time and Dose Dependent Manner" International Journal of Molecular Sciences 11, no. 12: 4843-4863. https://doi.org/10.3390/ijms11124743