1. Introduction
Investigations of electrostatic interactions, taking place on or in the vicinity of surfaces, are important for gaining a deeper understanding of interfacial dynamics. For example, enzymology studies have elucidated how enzymes can make beneficial use of a strong attractive electric field (produced by charges on the enzyme surface) to enhance enzyme-substrate association [
1,
2]. Differences in transport selectivity of ions through membrane-spanning proteins have also been attributed to distinct electrostatic properties of the funneled proteins [
3]. These examples resemble, to a large extent, how transport of charged particles (electrons and holes) is controlled in modern computer chips by intrinsic doping of charge densities and externally applied electric fields [
4]. Interfacial electrostatic interactions also play a key role during DNA transcription and replication. Recent findings show that week repulsion allows sliding of DNA binding proteins along the strain until attraction may occur at specific recognition sequences [
5]. Electrostatic interactions at interfaces also play a key role in the field of separation science,
i.e., chromatography or capillary electrophoresis [
6]; where the separation dynamics depend upon the analyte components, the mobile solution phase, and the surface properties of the packing or capillary support material. Given the key role of separation techniques for biomedical, pharmaceutical, and environmental analyses a large amount of techniques have been developed to characterize interfacial dynamics [
6].
Total Internal Reflection Fluorescence Microscopy (TIR-FM) is a suitable technique to study the mobility of fluorescent particles or molecules near surfaces. By exciting with evanescent laser excitation a very thin layer at the surface (typically about one hundred nanometers thick), one elegantly confines the probed volume and allows interfacial dynamic investigations [
7]. Using TIR-FM, the restricted motion of single organic dye molecules has been observed at fused-silica surfaces [
8]. The electrostatic contribution to adsorption on such surfaces has also been studied [
9]. TIR-FM has further been applied to investigate the restricted motion of single dye-labelled protein molecules and single intercalator-labelled DNA molecules at fused-silica surfaces at various pH and ionic strengths [
10,
11]. The technique has additionally been used to investigate the dynamic properties of fluorescent beads in the vicinity of bare and coated glass surfaces [
12–
14].
The motion of fluorescent probes at interfaces can further be analyzed by Fluorescence Correlation Spectroscopy (FCS) [
15]. The principle behind this analysis is the detection of fluorescence fluctuations from a small probe volume followed by statistical evaluations of the time-dependence of these fluctuations, brought about by the dynamics of individual fluorescent molecules [
16]. In its simplest form, the fluctuations are governed by molecules diffusing in and out of the probed volume [
17]. Directed movements, such as flow in and out of the probe volume, may also contribute to the fluorescence fluctuations [
18–
20]. Furthermore, chemical reaction kinetics, conformational changes, protonation reactions or photophysical processes, may also cause the fluorescence to fluctuate [
16,
17,
21–
23]. By use of FCS, it is in principle possible to deduce information about any dynamical process that manifests itself as a change in fluorescence intensity. To obtain the information, the fluorescence signal is analyzed in terms of correlation functions that give qualitative information of dynamic entities, such as, diffusion coefficients, chemical reaction rates, flow speeds, triplet-state kinetic rates,
etc. [
24,
25].
By combining FCS with evanescent laser excitation, known as Total Internal Reflection Fluorescence Correlation Spectroscopy (TIR-FCS), restricted motion of fluorescently labelled antibodies near phospholipid bilayers have been studied [
26,
27]. The restricted motion of fluorescently labelled vesicles diffusing near lipid membranes has also been investigated with TIR-FCS [
28]. The technique has additionally been used to investigate electrostatic contributions to adsorption kinetics of organic dye molecules in the vicinity of bare and coated silica surfaces [
29]. Evanescent laser excitation and fluorescence detection is either done through the same objective [
30], which is different to the more common system that uses prism-based total internal reflection excitation, and an additional objective for fluorescence detection [
15]. With the former method we have investigated diffusion of dianionic and cationic organic dyes molecules in pure aqueous solution at glass interfaces [
31,
32]. In addition, we have further applied it for investigation of DNA-hybridization, single enzymes kinetics, triplet-state dynamics, and protein and surfactant interactions at bare and coated glass surfaces [
33–
36].
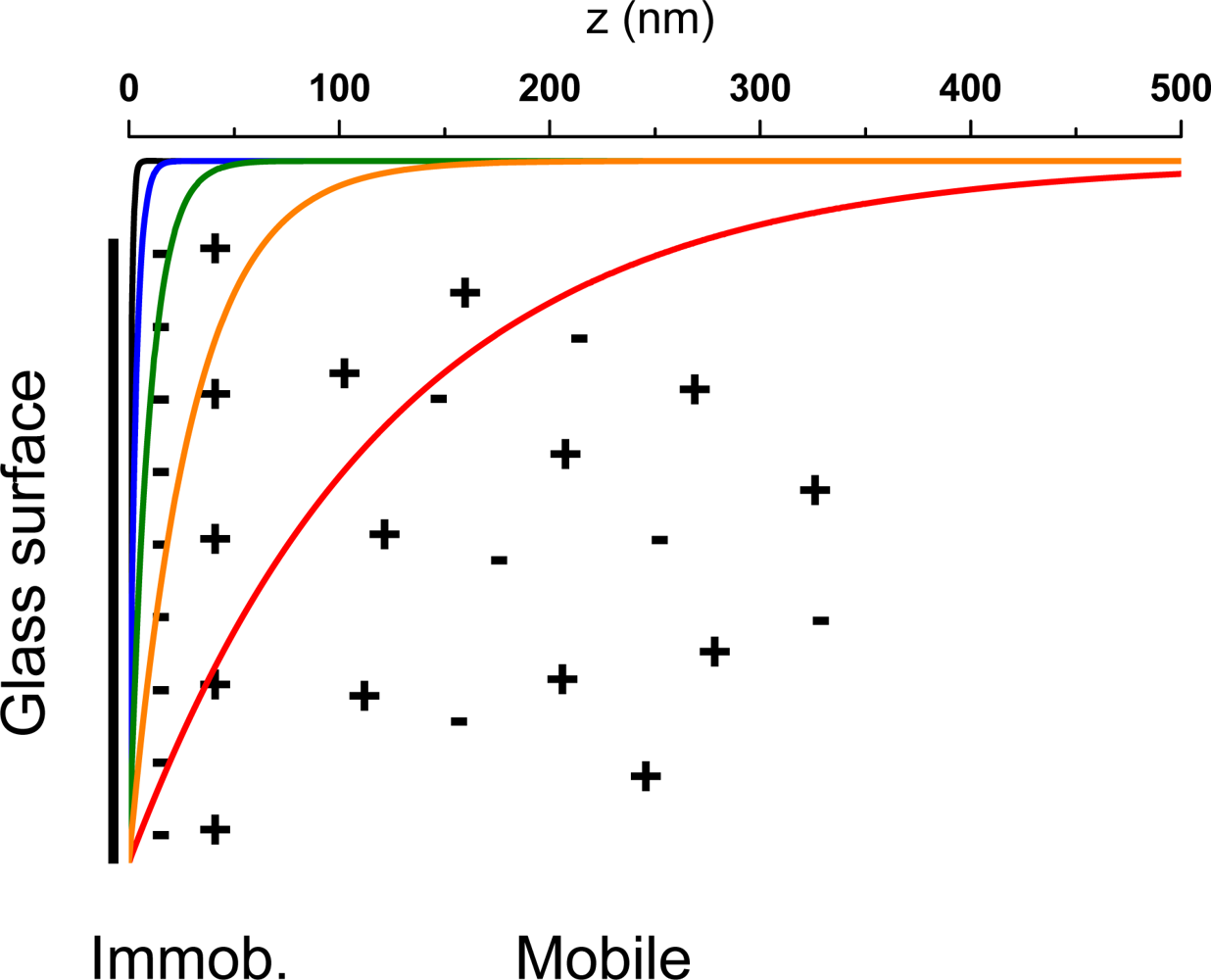
In this work, objective-based TIR-FCS is applied to investigate electrostatic interaction of fluorescent molecules with different charge (cationic, zwitterionic/neutral, and anionic/dianionic) and negatively charged dielectric glass surfaces, under different ionic strengths around physiological pH. The large information content available with TIR-FCS is used to deduce concentration changes, mobility variations, and photophysical kinetics parameter. As analyzed by means of the amplitude and time-evolution of the autocorrelation function, the fluorescent molecules experience electrostatic attraction or repulsion at the glass surface depending on their charges. Influences of the electrostatic interactions are also monitored through the triplet-state population and triplet relaxation time, including the amount of detected fluorescence or the count-rate-per-molecule parameter.
3. Results and Discussion
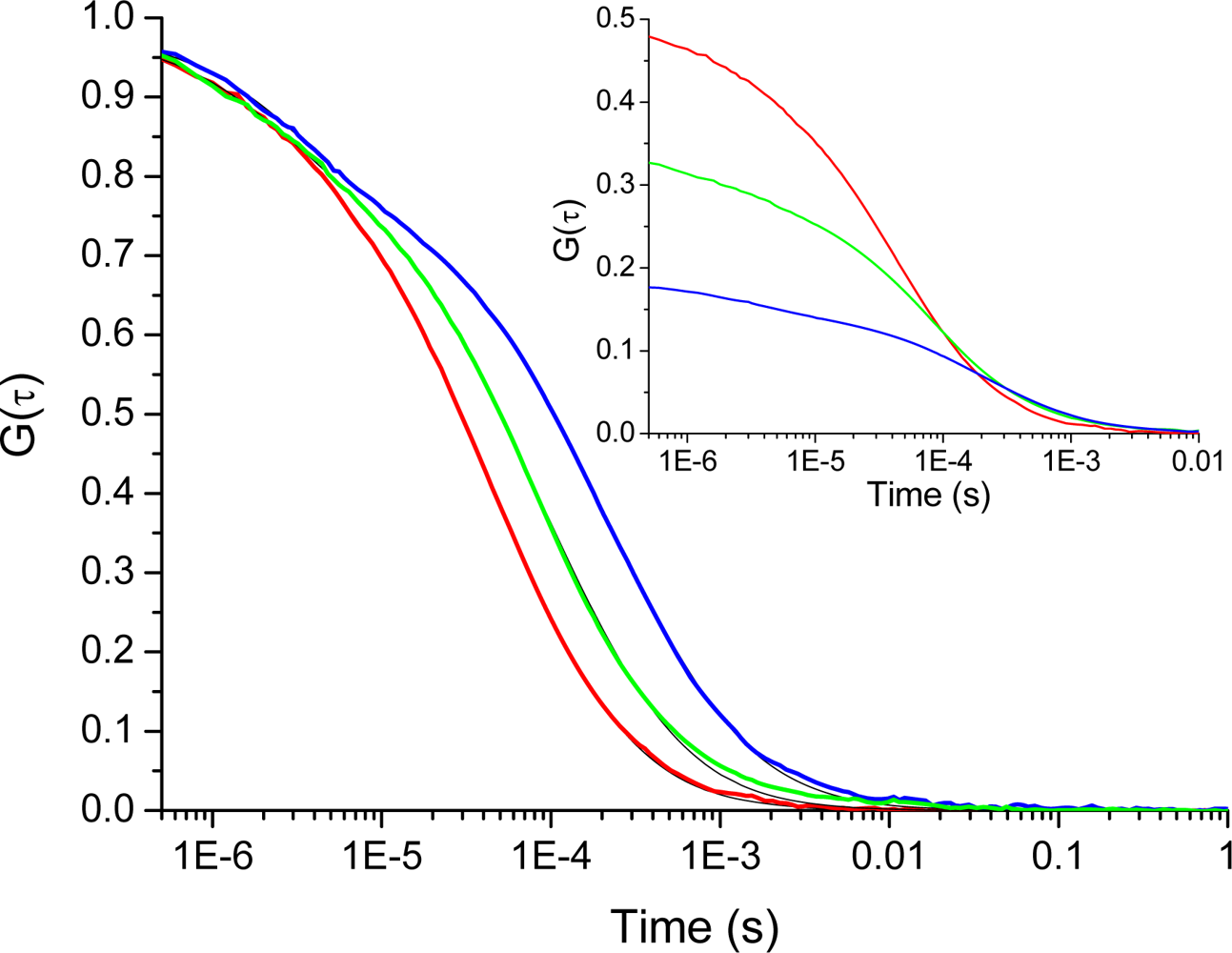
Figure 3 shows the TIR-FCS autocorrelation curves for cationic rhodamine 123 interacting with the negative glass surfaces at different ionic strengths. The influence of surface potential on the molecular motion manifests itself in an increased decay-time with decreasing ionic strength (
i.e., a less screened surface potential). The inset shows how the concentration of molecules, in the vicinity of the surfaces, increases as the ionic strength decreases from 100 mM to 0.1 mM. This is reflected in a decrease in the amplitude of the correlation curve. Both the shift in amplitude, as wells as the shift in decay-time of the correlation curves, clearly shows that the positively charged rhodamine 123 is attracted to the negatively charged dielectric surface. The small deviations seen at long times are most probably due to rare adsorption/desorption events not included in the present analysis.
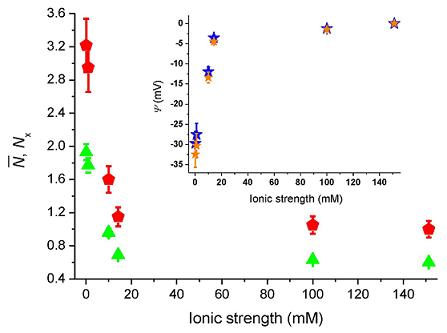
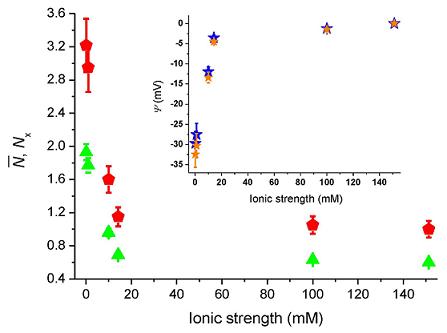
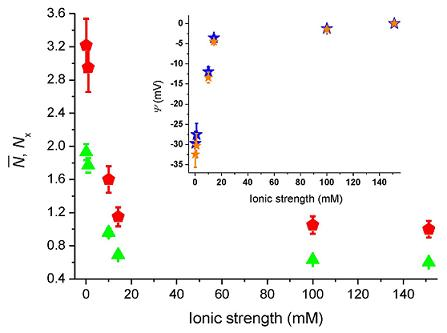
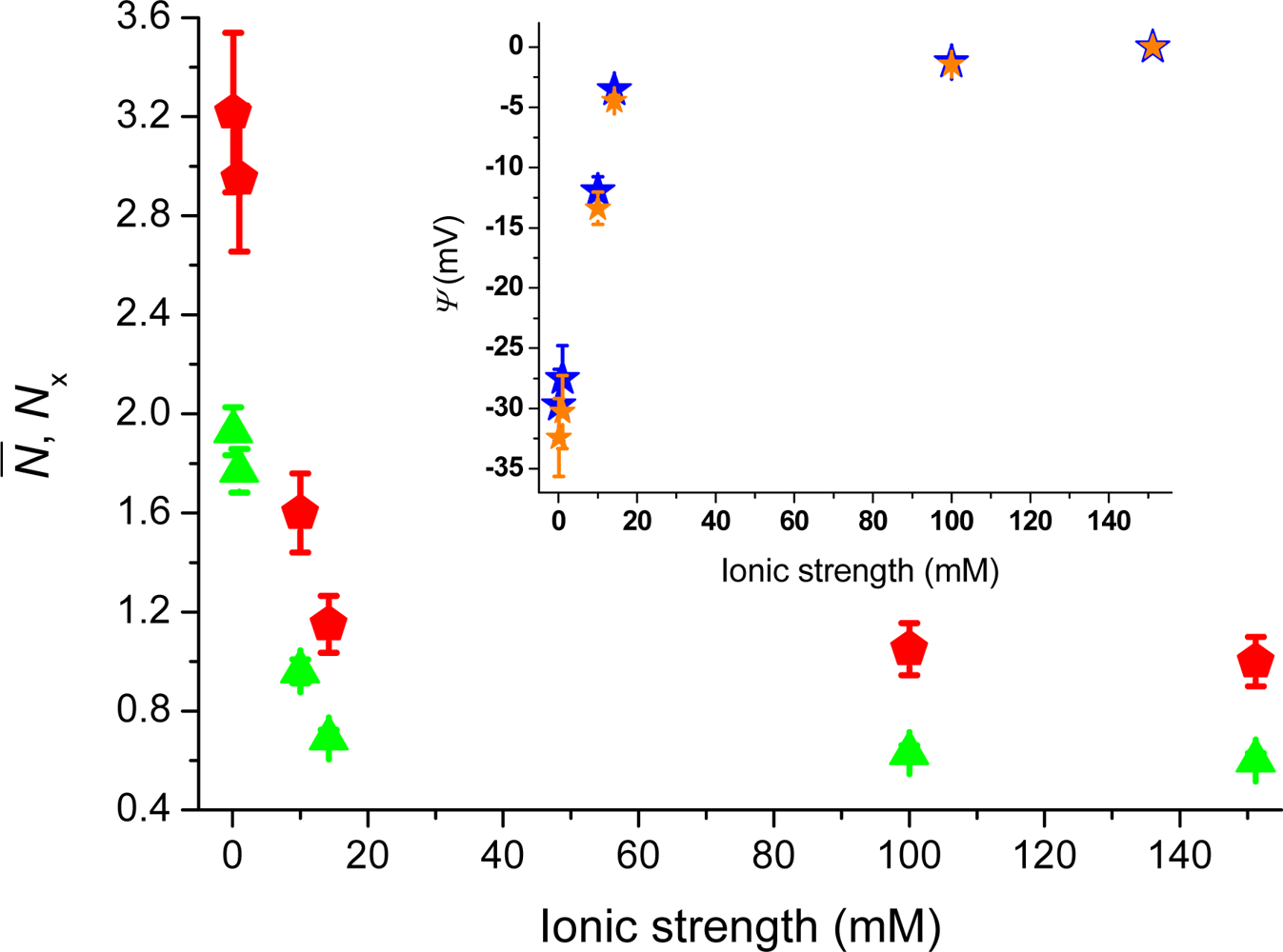
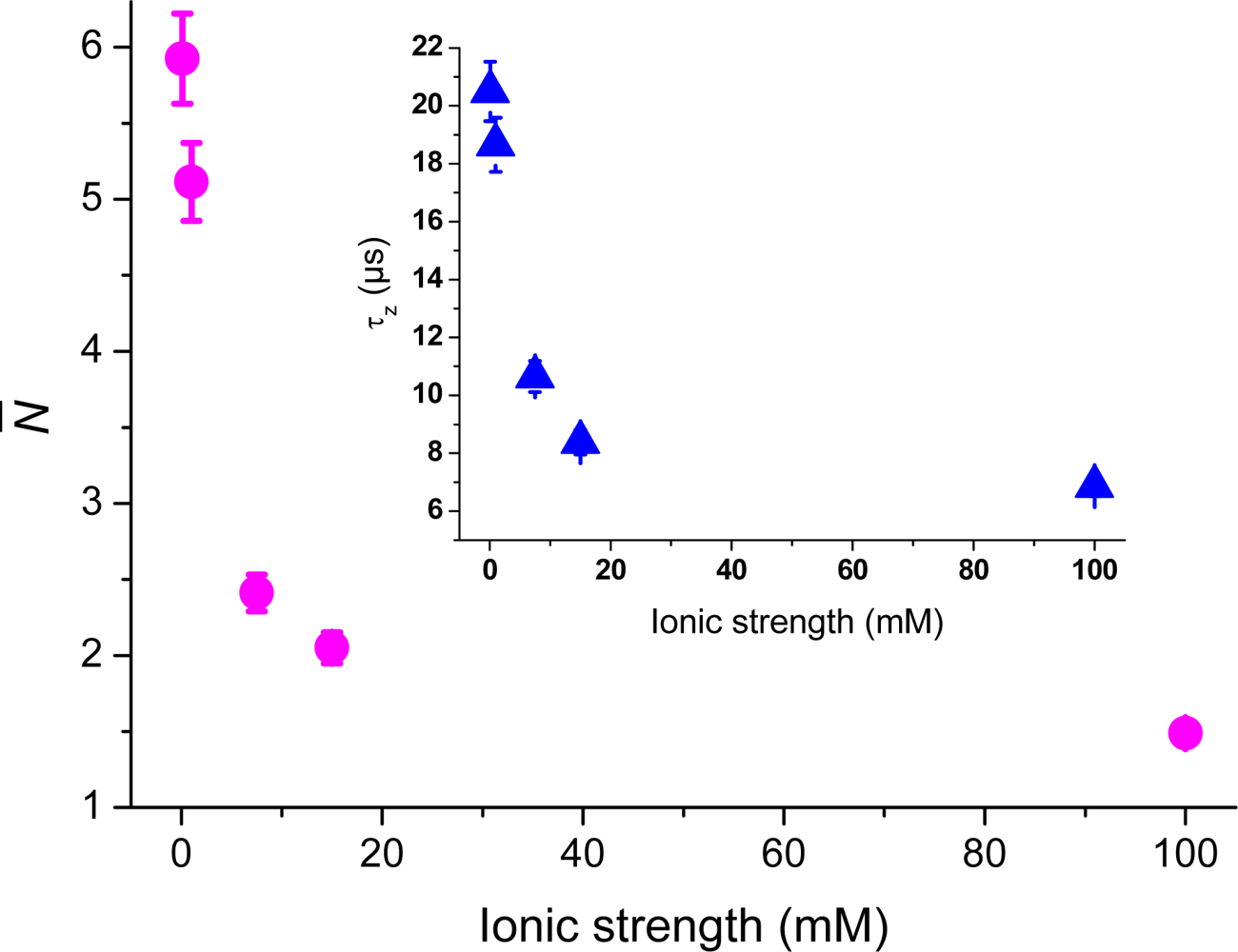
Figure 4 shows the number of molecules,
N̄, deduced from the correlation amplitudes together with the ratio of molecules,
Nx =
N̄ /
N̄B, used
via Equation (5) to evaluate the surface potential plotted in the inset,
i.e., Ψ(z) = Ψ = ζ. For an ionic strength of 0.1 mM, we deduce ζ~–30 ± 3 mV (pH = 6.6), with the bulk concentration,
N̄B, estimated from the data at 150 mM. The strength of the mean potential is slightly lower than values reported previously for glass surfaces. Investigations with TIR-FM on glass coverslips probed by fluorescent nanoparticles gave ζ~–44.4 mV (pH = 6.5) [
12]. TIR-FM studies on Pyrex glass gave ζ~–66 ± 8 mV (unknown pH) [
13]. For polished cover slips a value of ζ~–87 mV (pH = 6.5) was measured by electro-osmosis [
39]. As the literature values show, measured surface potentials depend on the kind of glasses used and on differences in preparation and cleaning protocols (wet and dry steps). The applied techniques also evaluate somewhat different parameters.
In electro-osmosis, the velocity of a liquid in response to an applied electric field in, for example, a capillary is used to measure the surface potential [
6]. In TIR-FM, the detected fluorescence intensity is used to estimate the position of fluorescent particles at the surface as a function of
z-position. In principle, this information is encoded in the detected fluorescence
via the exponential dependence of the evanescent excitation,
i.e.,
Equation (1), which magnifies small axial position changes into large fluorescence changes. However, as the fluorescence emission is highly non-isotropic at an interface [
37], the correlation between distances and detected fluorescence measurements can be biased [
12].
In TIR-FCS, the number of molecules at the interface is extracted through the amplitude of the correlation curve. The amplitude can likewise be biased by contributions of uncorrelated background signals, consisting for example of back-reflected and scattered laser light. Contribution of such background signals decreases the amplitude of the correlation curve and leads to an overestimation of the number of molecules in the probed volume,
i.e.,
N̄. To correct for bias in the amplitude, the “true” number of molecules,
N0, may be extracted using the following expression:
N̄0 =
N̄ (1 +
B/
F)
−2, where
B is the background signal that can be measured for a sample of pure water, and
F is the detected fluorescence signal [
24,
40]. Incorporating this correction, with a
B/
F ratio that varied from 0.07 to 0.02 as the ionic strength increased from 0.1 mM to 150 mM, leads to a modest change of the surface potential from −29.7 mV to −32.4 mV at 0.1 mM (cf. blue and orange stars in the inset of
Figure 4). Note that when error-bars are not visible, the errors are contained in the size of the markers.
This unbiased value can further be compared to the numerical analysis of expression (7), which incorporates the effect of the electrostatic concentration profile to the correlation amplitude. Applying this expression to the measured values of the amplitude,
i.e.,
G(0) =
γ[
N̄×(1–
T)]
−1, yields a surface potential of about −94 ± 8 mV at an ionic strength of 0.1 mM, which is higher than that deduced directly
via Equation (5). The bulk concentration
N̄B =
Veff C̄B was extracted at 150 mM ionic strength, and the axial extent of the detection volume,
h, was deduced by the axial passage time
τz,
via, assuming D~4.1 × 10
−10 m
2s
−1 [
41] (cf.
Figure 5). The numerically extracted surface potential is slightly higher than the cited literature values [
12,
13], but agrees fairly well with values for mechanically polished glass [
39]. This might be explained as an effect of our oxygen plasma etching step, which probably increases the density of attracting silanol groups on the glass surface.
Furthermore, the attraction of cationic rhodamine 123 towards the negative glass surface manifests itself in a change in the main decay-time of the correlation curves (cf.
Figure 3).
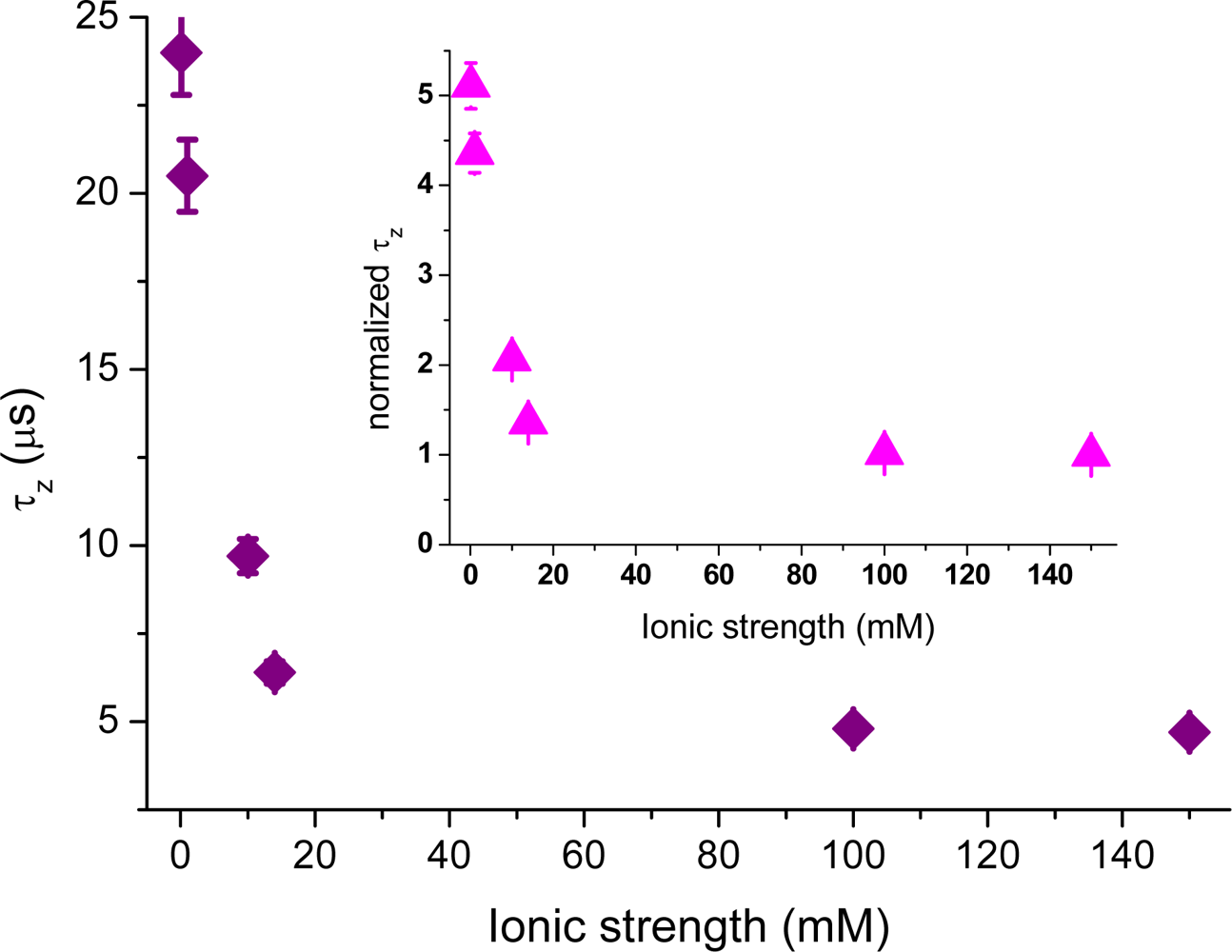
Figure 5 shows the dependence of the average axial passage-time,
τz, on the ionic strength, deduced by fitting the autocorrelation data with
Equation (4). A large increase in the axial passage-time through the probe volume located at the glass surface is observed with decreasing ionic strength. The same increase as seen at 0.1 mM ionic strength was also seen in (pure) aqueous solution having pH = 6.6 (data not shown). By normalizing the axial passage-time, shown in
Figure 5, with the passage time given for a fully screened surface a plot resembling that of the increased concentration profile,
Nx, shown in
Figure 4 is generated. The inset shows that the normalized passage time of the cationic molecules can be prolonged up to a factor of five, meaning that they spend a considerably longer time in the probed volume at low ionic strengths. From the data shown in
Figure 5 it should therefore in principle be possible to deduce the surface potential, through a model of charged particle diffusing in an electric potential as introduced by Smoluchowski [
42]. However, the nonlinearity of the potential makes it difficult to derivate an analytical expression for the autocorrelation function. Fitting of the electrostatic affected axial passage time is hence performed (so far) with an expression assuming free diffusion,
i.e.,
Equation (4). In previous studies, the diffusion coefficient of anionic antibodies at cationic surface membranes was seen to be larger at low ionic strengths and smaller at high ionic strengths [
26], which is opposite to what is seen in
Figure 5. Note that the inverse of the axial passage-time is proportional to the diffusion coefficient (
i.e.,
τz ∝ 1/
D). This opposite relation could be explained by influences from hydrodynamic interaction between the antibody and the membrane surface [
27]. Interfacial interaction on coated surfaces, is therefore often a somewhat more complex dynamic process, and the use of different coatings may actually amplify both hydrophobic [
13,
29], as well electrostatic interactions [
13,
39]. Also the hydrophobicity of the probe molecules may influence the surface dynamics [
29], which leads to chemical interaction occurring simultaneously with charge driven electrostatic contributions.
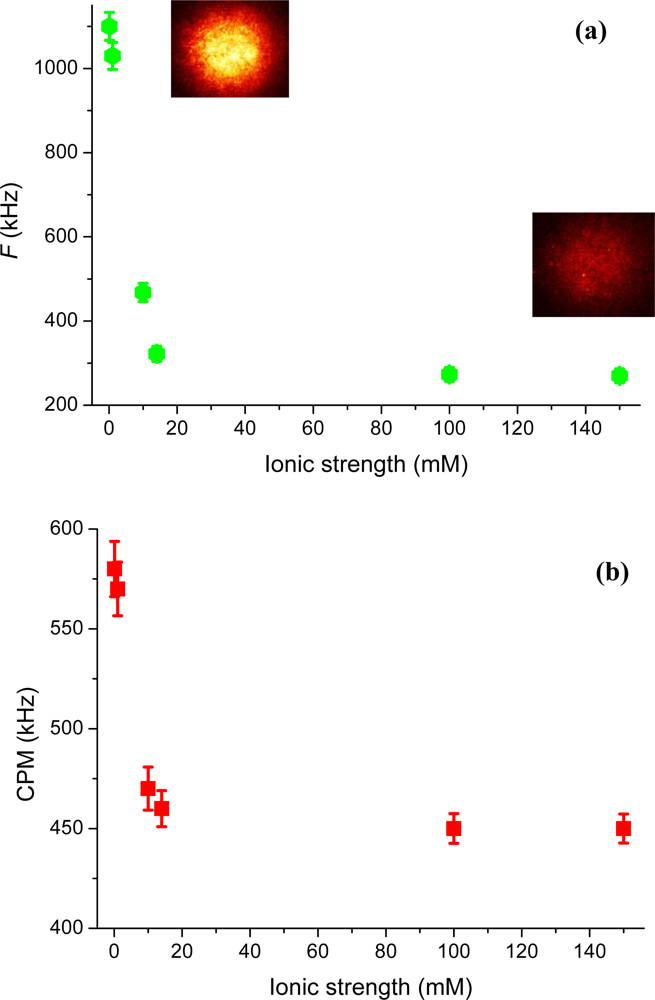
The electrostatic interaction at our uncoated glass surface can also be investigated through the detected fluorescence on the SPAD detector, or on the CCD camera.
Figure 6a shows the detected fluorescence,
F, as a function of ionic strength. Images of the detected fluorescence for rhodamine 123, monitored on the CCD at 0.1 mM and 150 mM ionic strength, are also shown. The deduced count-rate-per-molecule (in kHz), calculated as CPM =
F/
N̄, is further shown in
Figure 6b. A comparison between the detected fluorescence and the CPM reveals that the former increases by a factor of four, where as the latter only increases about 1.3 times when the ionic strength is lowered. This is understandable as more (attracted) cationic molecules contribute to the signal at low ionic strength and are adding further to the detected signal through the larger (on average) experienced excitation irradiance (
i.e., being closer to the surface). However, the added signal does not increase linear with the excitation irradiance as saturation of the fluorescence emission may occur.
Emission rates of fluorescent molecules are largely determined by the excitation irradiance (
i.e., W/cm
2) at which saturation of the singlet-state is reached, and this in turn depends on the mean occupancy of the long-lived triplet state [
23]. While the fluorescence lifetime of the singlet-state of an organic dye molecule typically is in the nanosecond range, the lifetimes of the triplet state is in the microsecond to millisecond range, depending on the environment of the molecule. Consequently, the triplet state has ~10
3–10
6 more time to interact with the immediate environment, compared to the fluorescent singlet state. The kinetics of the triplet state thus represents an additional dimension of information, which can respond and change considerably due to small changes in the microenvironment, e.g., accessibility of quencher molecules or solvent viscosity [
23].
In this work, the influence of the presence of the glass surface on the triplet-state dynamics is also studied.
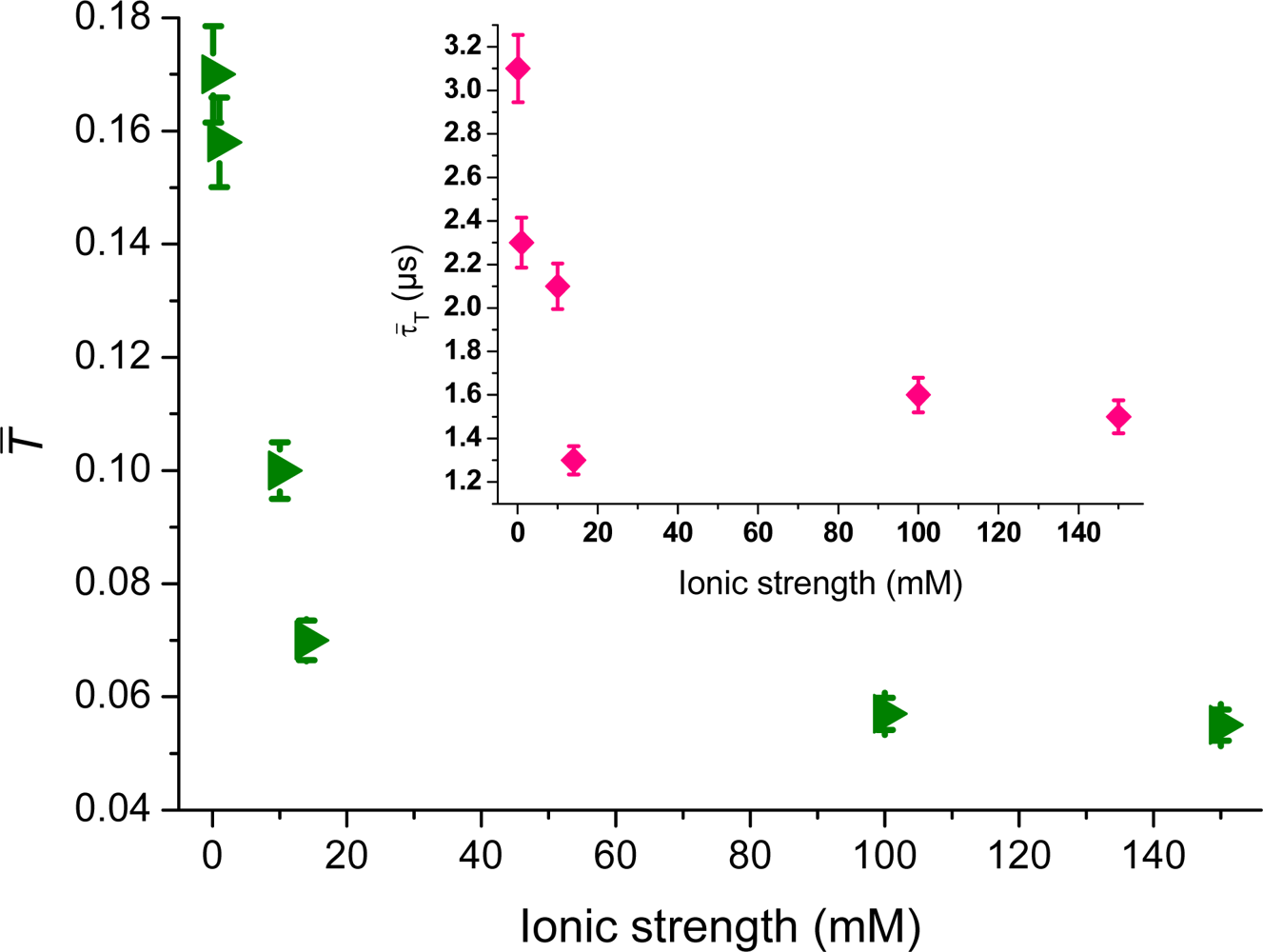
Figure 7, shows the changes of the TIR-FCS deduced triplet amplitude,
T̄, as function of the ionic strength for rhodamine 123. This is deduced by fitting
Equation (4) to the autocorrelation data shown in
Figure 3. When decreasing the ionic strength, the triplet-state population is increased in response to the increased electrostatic attraction, shifting more cationic molecules towards the larger excitation irradiances at the surface. The inset shows the triplet relaxation time,
τ̄T, that shows a similar trend as the triplet-amplitude. This is in agreement with the population kinetics of the triplet state, where longer
τ̄T values generate larger
T̄ values. Triplet-state kinetics of organic dye molecules in the vicinity of glass surfaces has been investigated previously at high ionic strengths (
i.e., fully screened surfaces) [
36]. Extracting the triplet-state kinetic rates from the data on unscreened surfaces should in principle be possible, but is beyond the scope of this work. The non-isotropic concentration variation, due to the electrostatic concentration profile, then needs to be included in that rate analysis.
The examples above show the potential TIR-FCS has for investigating electrostatic interaction dynamics of single fluorescent molecules at interfaces.
Figures 8–
10 show additional examples of this for cationic rhodamine 6G, anionic/dianionic fluorescein, zwitterionic rhodamine 110, and neutral ATTO 488, monitored at various ionic strengths at negatively charged glass surfaces. As can be seen in
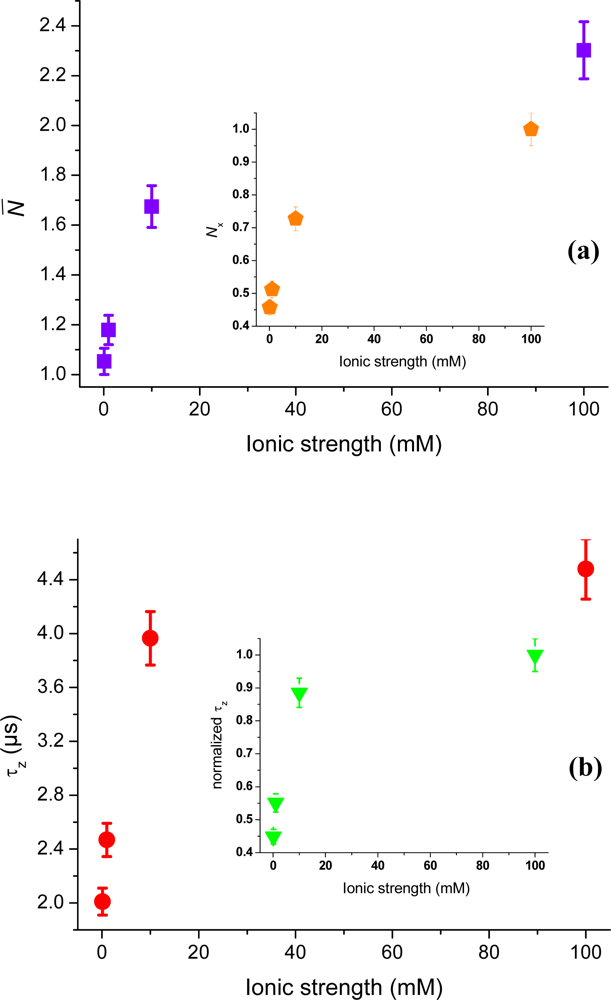
Figure 8, cationic rhodamine 6G shows a similar increase in number of molecules as cationic rhodamine 123 (cf.
Figure 4). The values are corrected for background contributions and all data points are obtained for molecules in aqueous solution at ionic strengths between 0.1 mM and 100 mM (pH = 6.6). Assuming that the surplus of rhodamine 6G molecules at the interface is due to electrostatic attraction,
Equation (5) yields a surface potential of about −35 ± 4 mV (pH = 6.6) at 0.1 mM ionic strength. The bulk concentration was here approximated with the number of molecules at 100 mM ionic strength. Applying expression (7) to the Rh6G values a surface potential of about −100 ± 9 mV at 0.1 mM ionic strength is deduced, which is similar to the cationic Rh123 values.
The reason for the slightly higher estimation for the potential might be the larger hydrophobicity of rhodamine 6G [
29]. A comparison at 100 mM ionic strength give
NRh6G = 1.48 and
NRh123 = 0.52, which is relatively well in agreement with the differences in concentrations. At 0.1 mM ionic strength the background corrected values are
NRh6G = 5.93 compared to
NRh123 = 1.86, which gives a difference of about 3.2 for a sample concentration difference of only 2.5. The calculated strength of the surface potential is therefore probably biased in the case of rhodamine 6G. The inset of
Figure 8 shows the axial passage-time,
τz, of rhodamine 6G, which indicates strong restricted motion with decreasing ionic strength. Again, a comparison to the passage time of rhodamine 123 reveals the same main trend, and where the small differences seen are probably due to different hydrophobicity of the two fluorophores. The difference is partly explained by Rh6G’s higher molecular mass, which contributes a factor of 1.08 to the difference of 1.4 seen in axial passages times at 100 mM ionic strength. Additional surface investigations with a combination of techniques (single particle tracking, confocal FCS, TIR-FCS, TIR-FM,
etc.) are probably needed to disentangle the full complexity of this interfacial dynamics [
9,
26,
27,
29].
Switching from cationic fluorescent molecules (
q = +1) to negatively charged fluorescein instead (anionic/dianionic,
q = −1 to −2), allows probing contribution of repulsion at the glass interface [
10,
13].
Figure 9a shows background corrected number of fluorescein molecules in the vicinity of the surface extracted from TIR-FCS measurements (data not shown). Compared to the attraction of cationic dyes, the concentration is now decreasing as the ionic strength decreases. The deduced concentration ratio of molecules,
Nx, due to the repulsion is shown in the inset. As the Debye-length of the repulsion increases, the ratio decreases over two times, meaning that less than half the number of molecules is located within the electric double layer at low compared to high ionic strengths. In addition to the changes in number of molecules, the axial passage-time,
τz, and the normalized axial passage-times are also shown in
Figure 9b. The results also give a clear indication of an electrostatic interaction repelling anionic/dianionic molecules from the negative glass surface. This repellent interaction forces fluorescein molecules to dwell mainly in the outer parts of the observation volume, resulting in a decreased axial passage-time at lower ionic strengths (
i.e., at a less screened surface).
Applying the fluorescein data to
Equation (5) allows us to estimate the surface potential, which becomes about −13 ± 2 mV (pH = 6.6) at an ionic strength of 0.1 mM, when assuming a charge of
q = −1.5. This mimics a mixture of the protonated and deprotonated form of fluorescein, HFl
− ←→ Fl
2− (pKa = 6.4) at this pH [
22]. Assuming further that the protonated anionic form is practically nonfluorescent (meaning it is only the dianionic form that contributes to the TIR-FCS data), the surface potential then becomes about −10 ± 2 mV. Applying expression (7) to the fluorescein values (assuming
q = 2) yields a surface potential of about −40 ± 4 mV at 0.1 mM ionic strength. The reason for the lower deduced surface potential might be due to a remaining electrostatic interactions even at 100 mM ionic strength, also seen by TIR-FM methods [
10,
11]. This decreases the number of molecules at the interface, and the ratio
N̄ /
N̄B, leading to extraction of a lower surface potential
via Equation (5). The bulk value should therefore have been measured at a higher ionic strength.
To circumvent the problems of bulk concentration estimates, a measurement of
N̄B in a confocal FCS mode (with a known probe volume) could be done before switching to surface based TIR-FCS investigations. This approach has recently been applied to studies of the restricted motion of fluorescently labelled vesicles diffusing near planar membranes [
28]. In that study it was also seen that the negatively charged probes experienced a repulsive force from negative surfaces. Deviation from theory was however seen at low ionic strengths, where the electrostatic decay lengths start to approach the size of the evanescent excitation profile. A pool of fluorescent probes might then be located outside the TIR-FCS detection volume, which leads to an underestimation of the measured concentrations, as pointed out above.
As a final example, neutral organic dyes (ATTO 488 and rhodamine 110,
q = ±0) were also applied to probe the negative glass surface. ATTO 488 is uncharged while rhodamine 110 is zwitterionic and has at neutral pH an unprotonated carboxyl group carrying a negative charge and an amino group carrying a positive charge.
Figure 10a shows the number of neutral probe molecules (corrected for background contributions) in the vicinity of the surface. As no main electrostatic charge-charge interactions are present, the measured concentration of both dyes stays constant for all ionic strengths. This is also true for the in
Figure 10b shown axial passage-times. Neutral fluorescent molecules can therefore be used as calibration standards, as they basically show no electrostatic attraction or repulsion to charged surfaces. The small variations seen come from noise or measurement error. A closer look at the data for rhodamine 110 reveals, however, that the axial passage-time is somewhat higher than expected compared to ATTO 488, which has a higher molecular mass. The larger than expected values for the rhodamine 110 may reflect some hydrophobic interactions or very weak remaining electrostatic influences following from its zwitterionic character. Again, further studies are needed to investigate the full complexity of that interfacial interactions dynamics.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









