Light Dose is a Limiting Factor to Maintain Cell Viability in Fluorescence Microscopy and Single Molecule Detection


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Test Samples
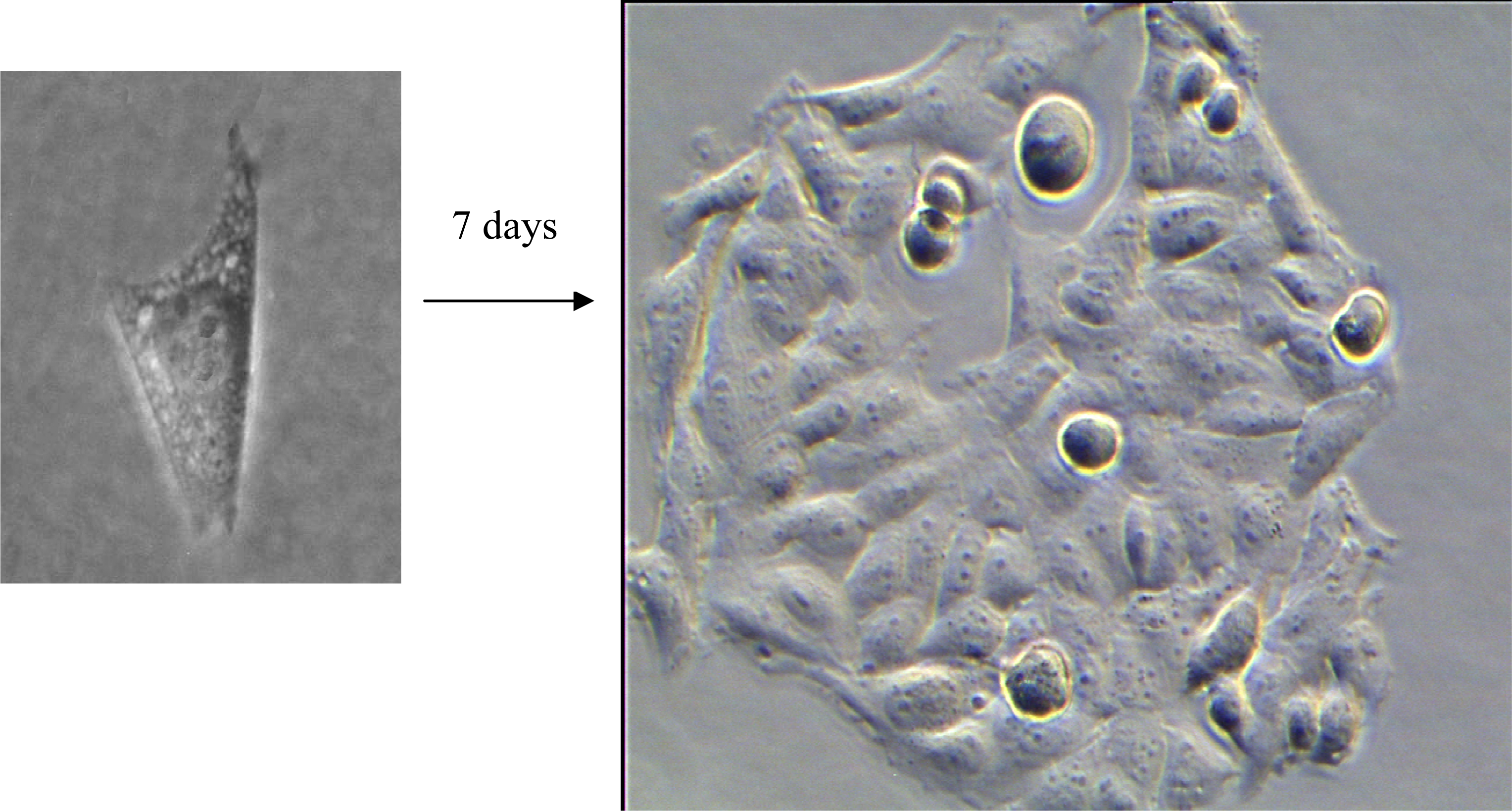
2.2. Colony Formation
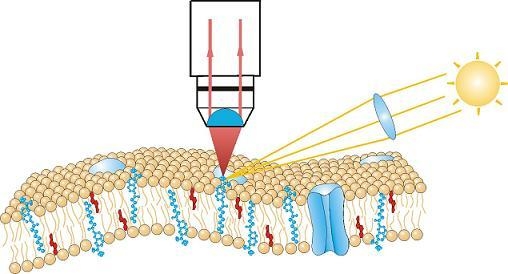
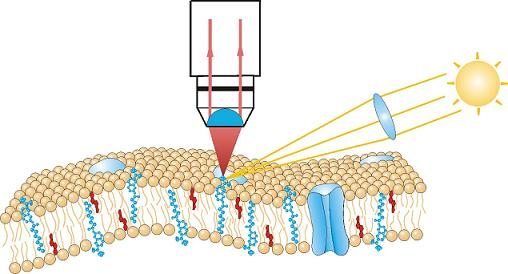
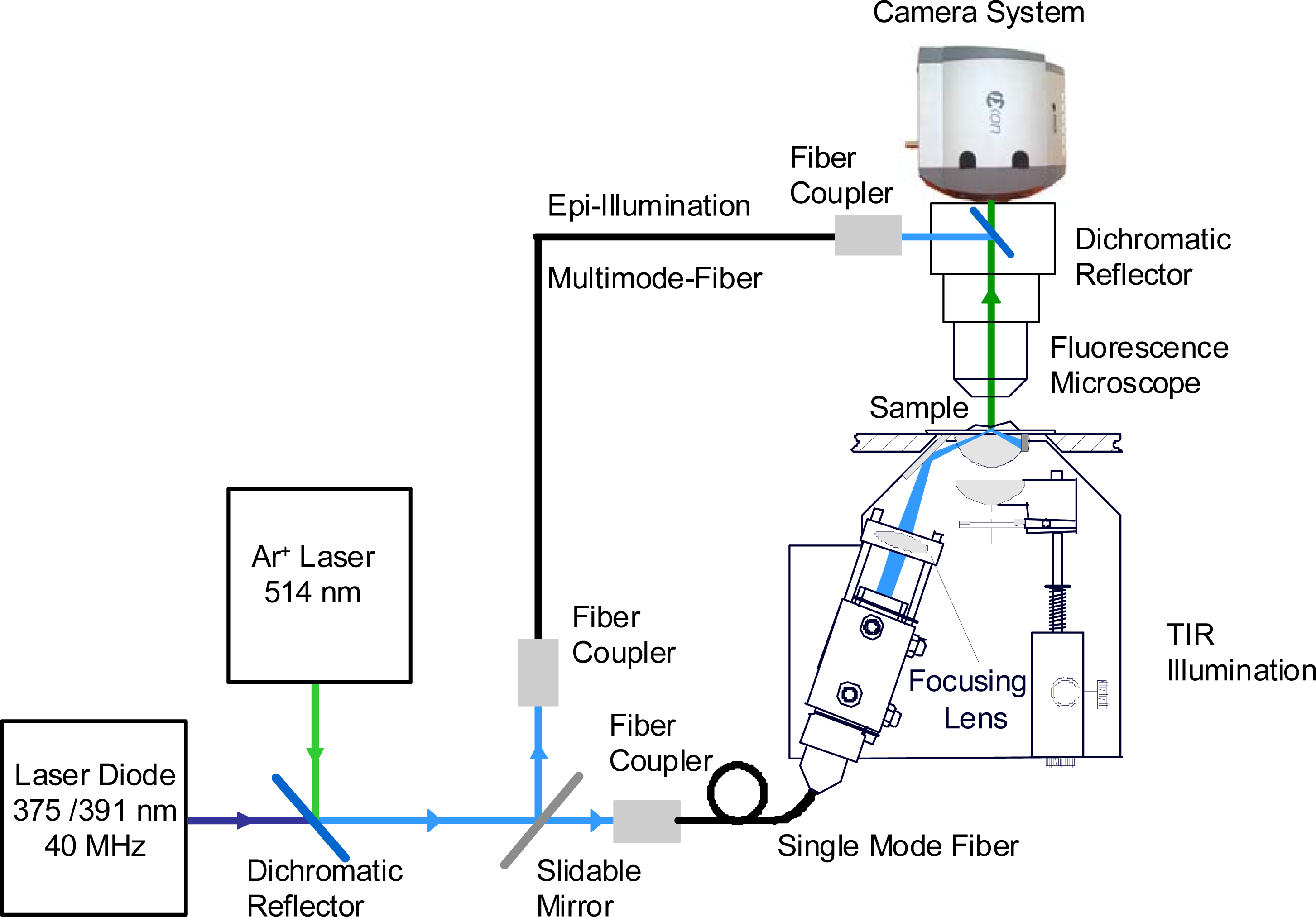
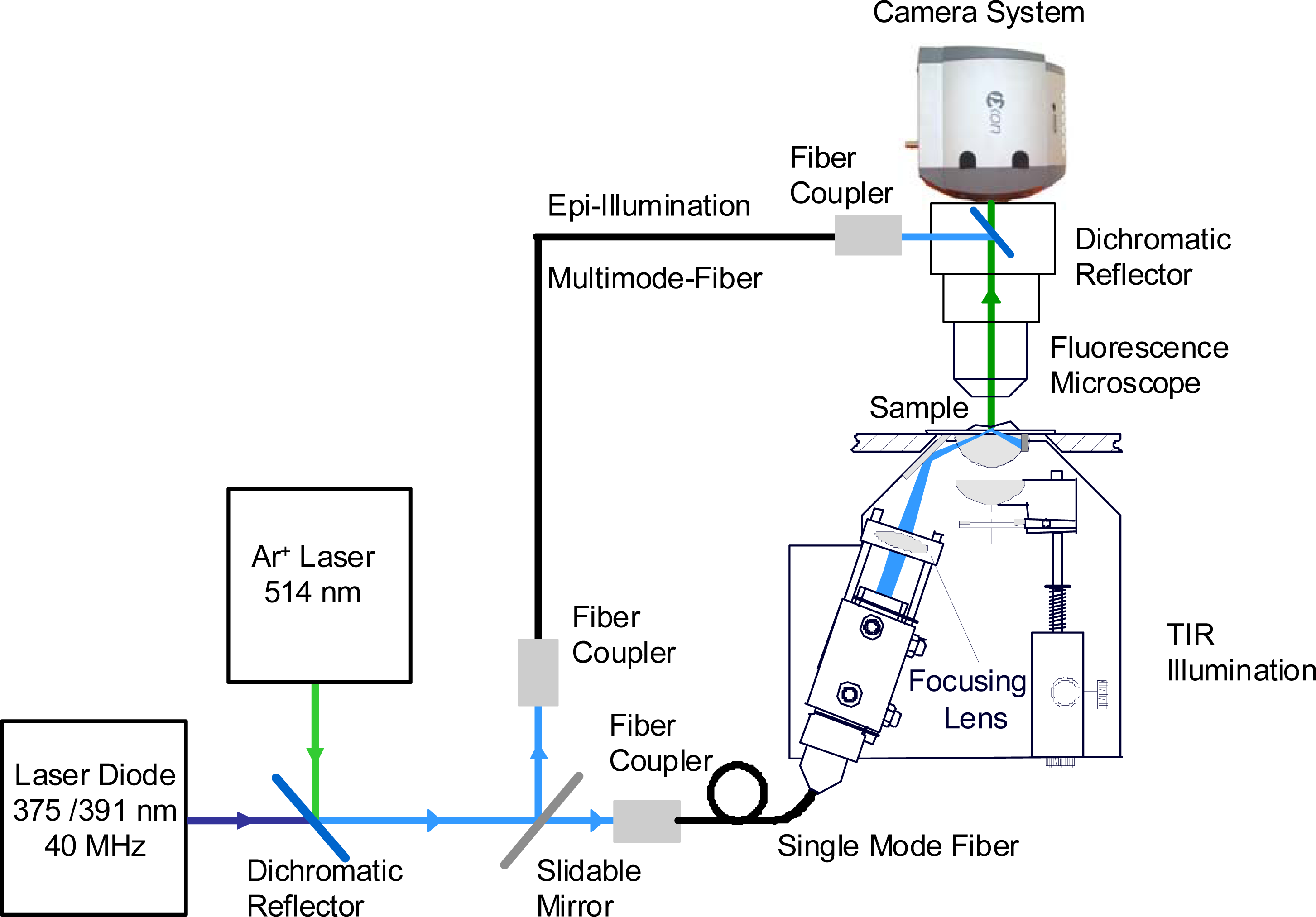
2.3. Fluorescence Microscopy
3. Results
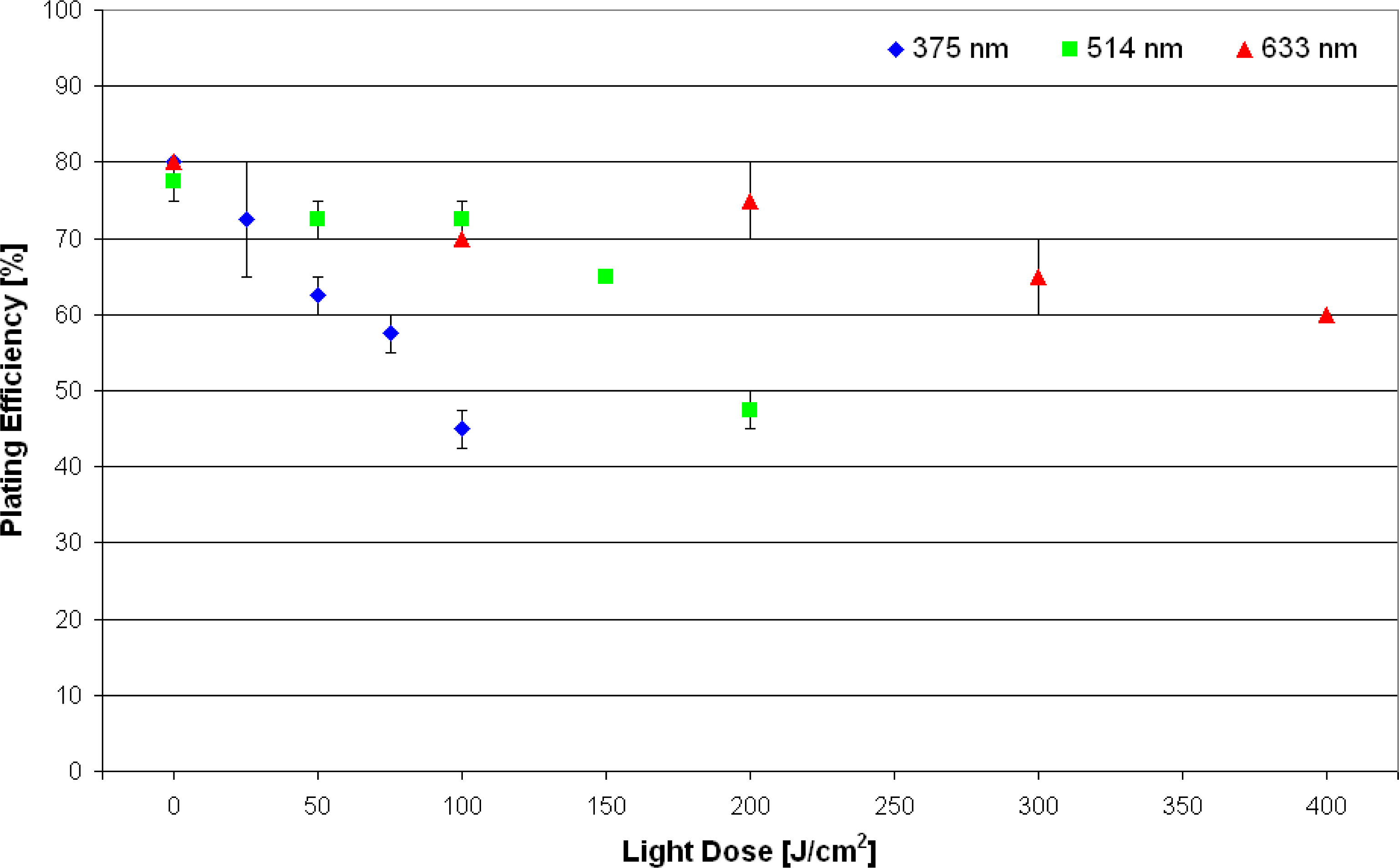
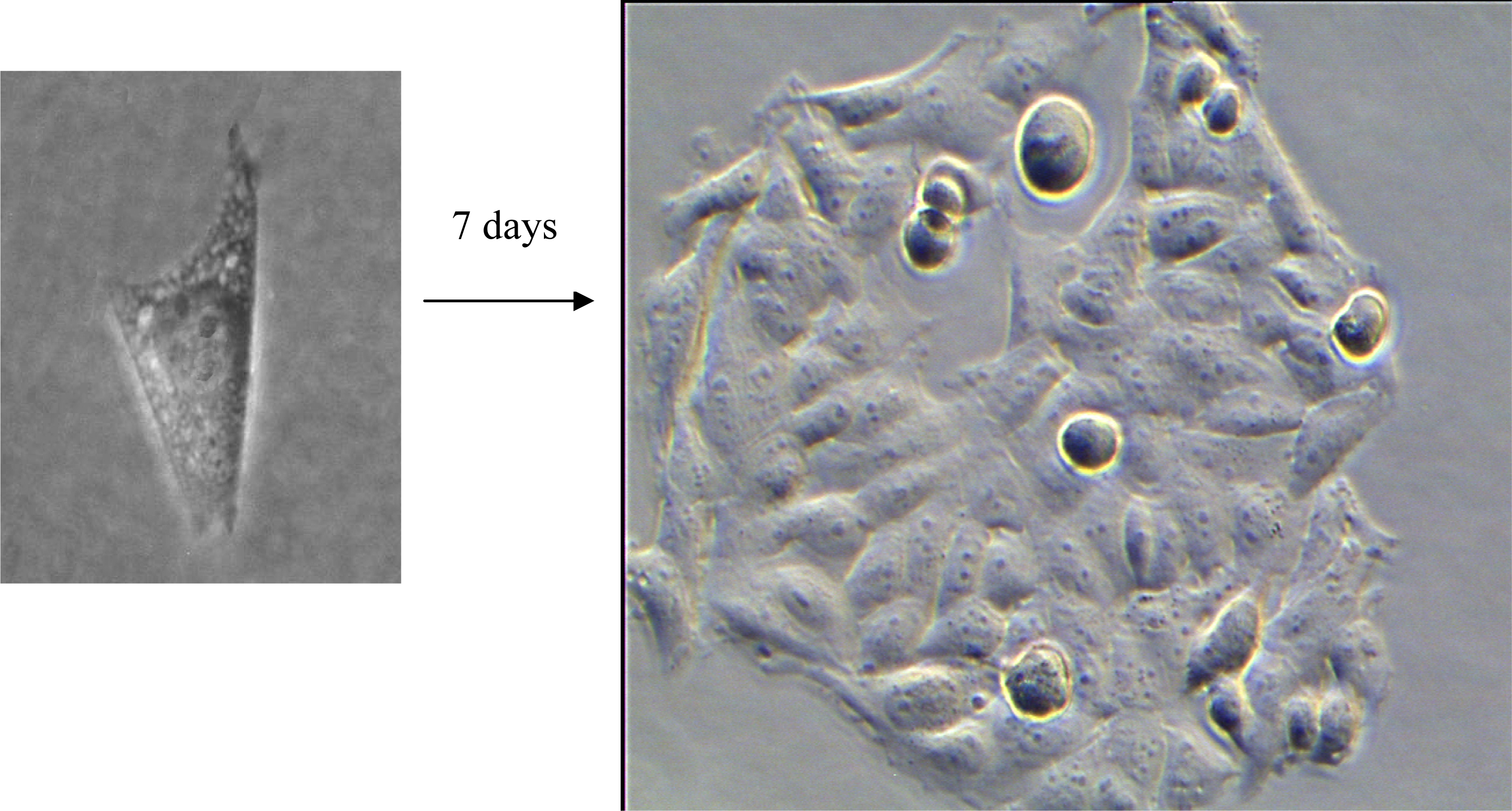
3.1. Colony Formation
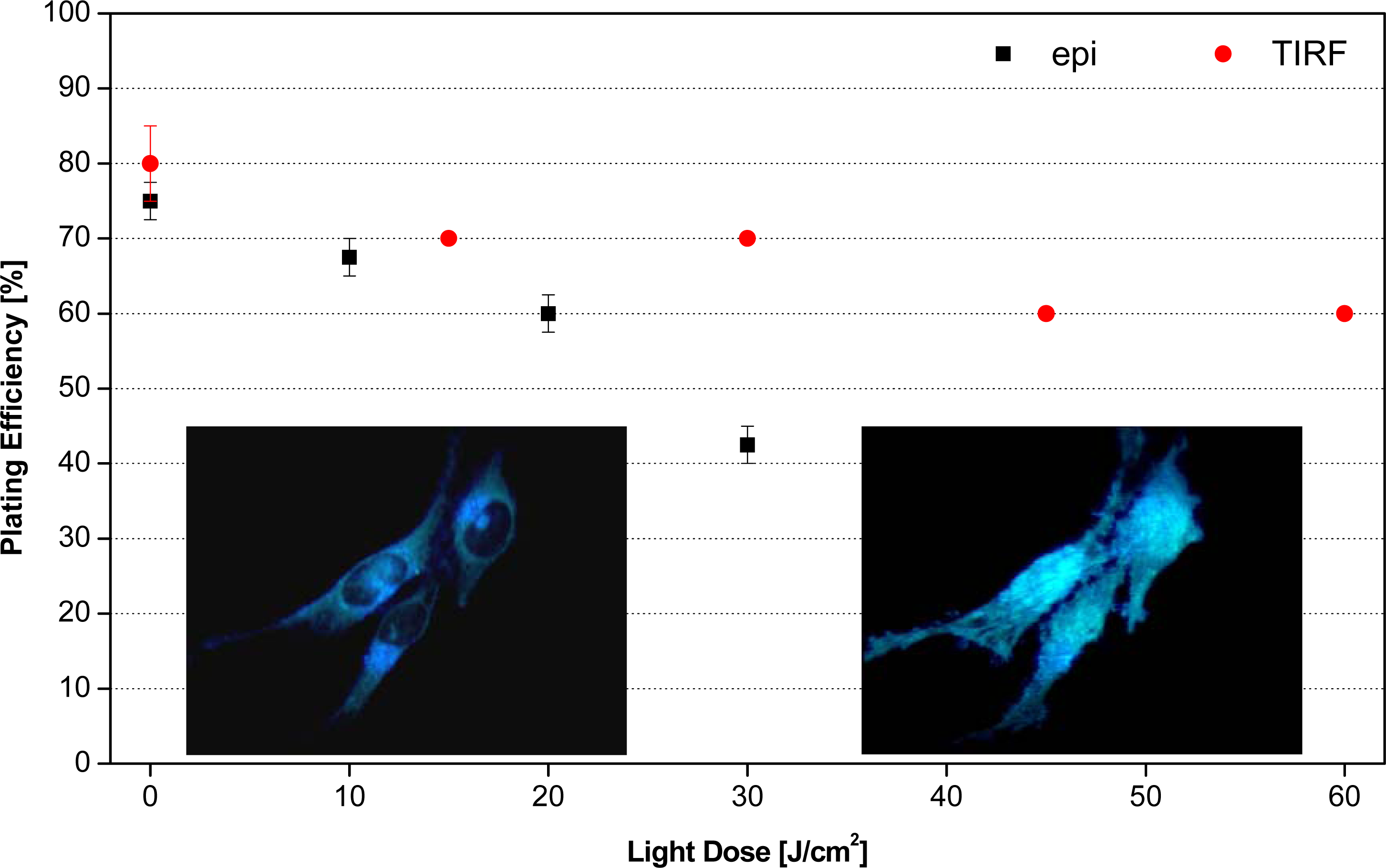
3.2. Fluorescence Microscopy of Living Cells
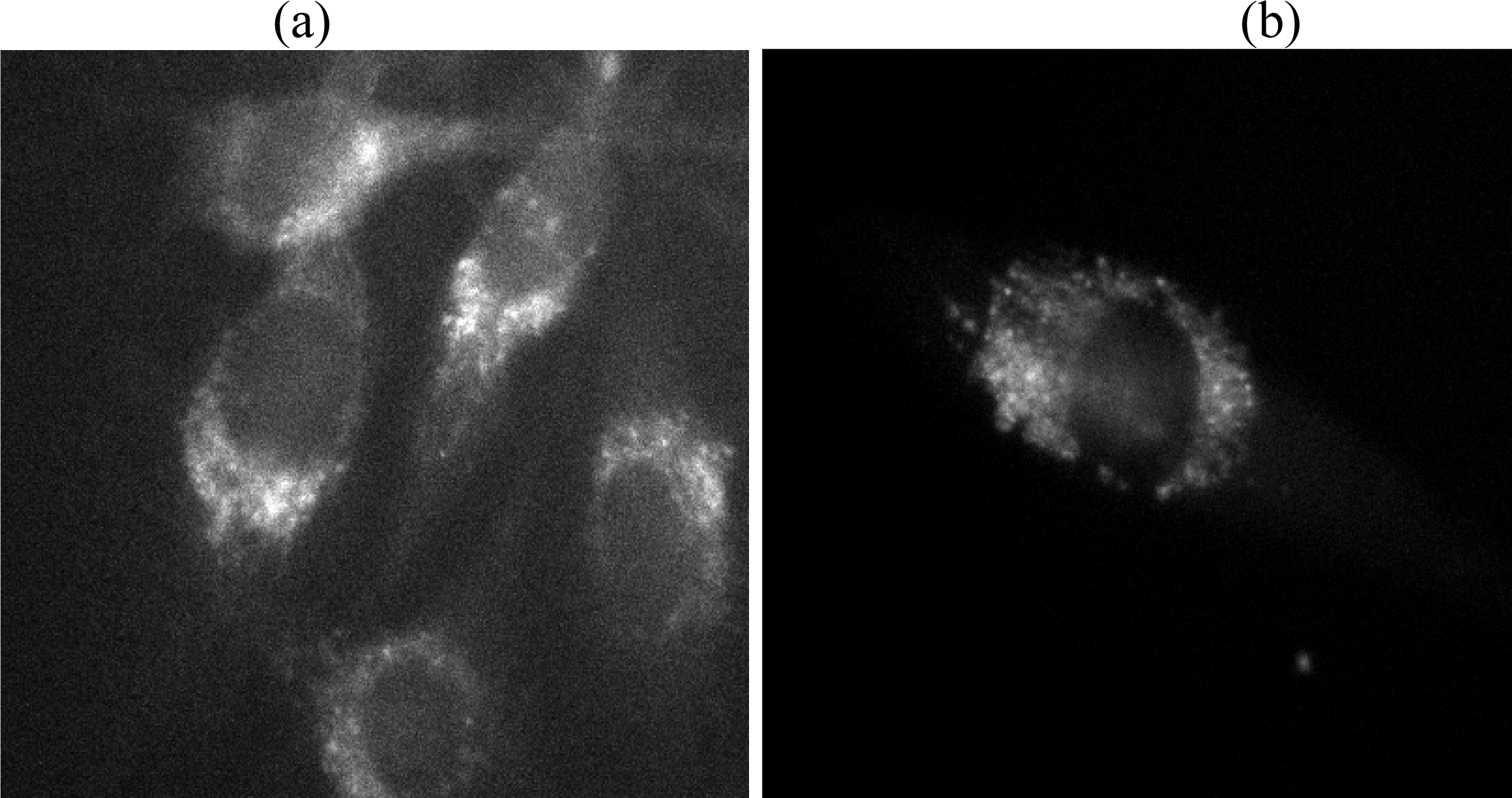
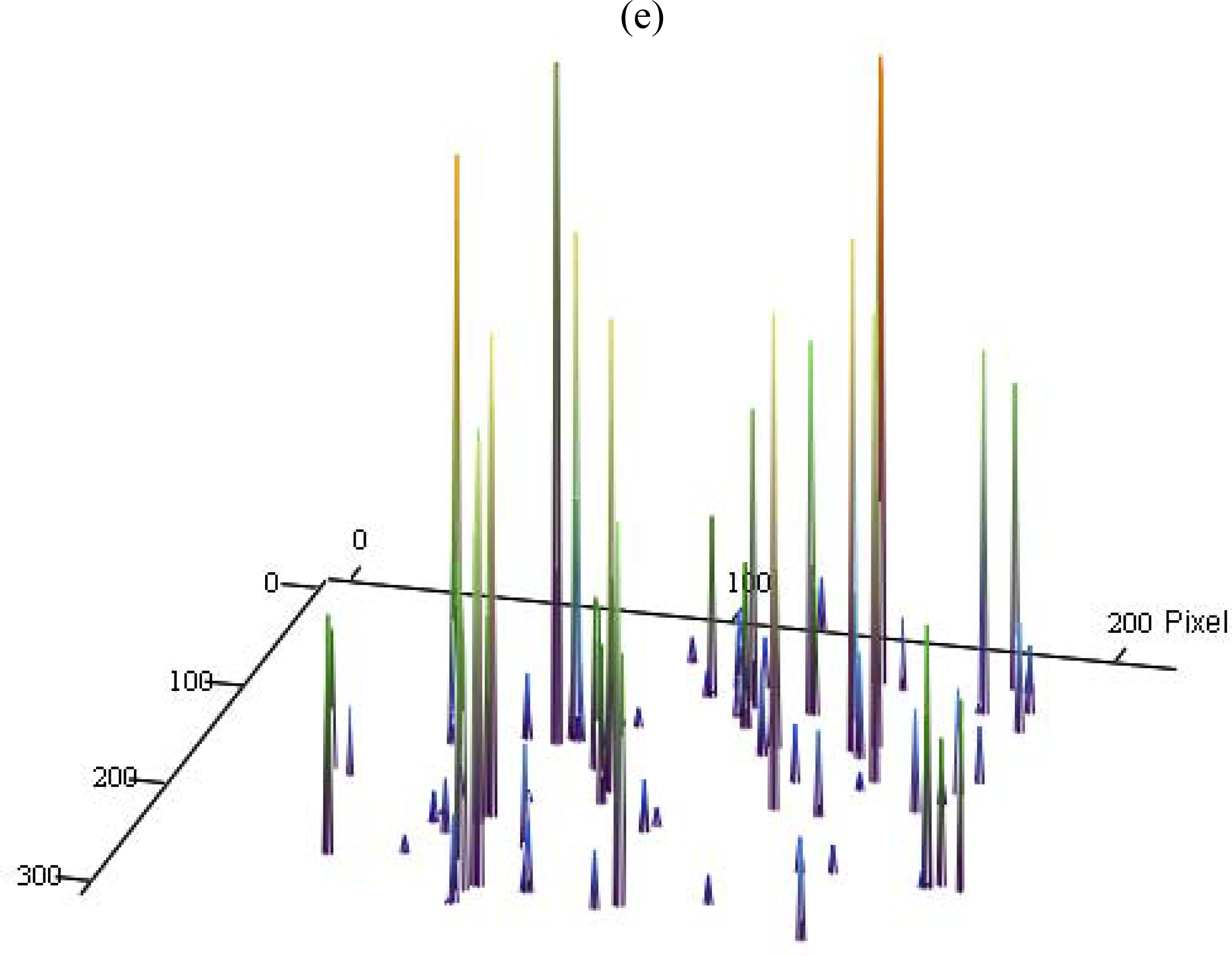
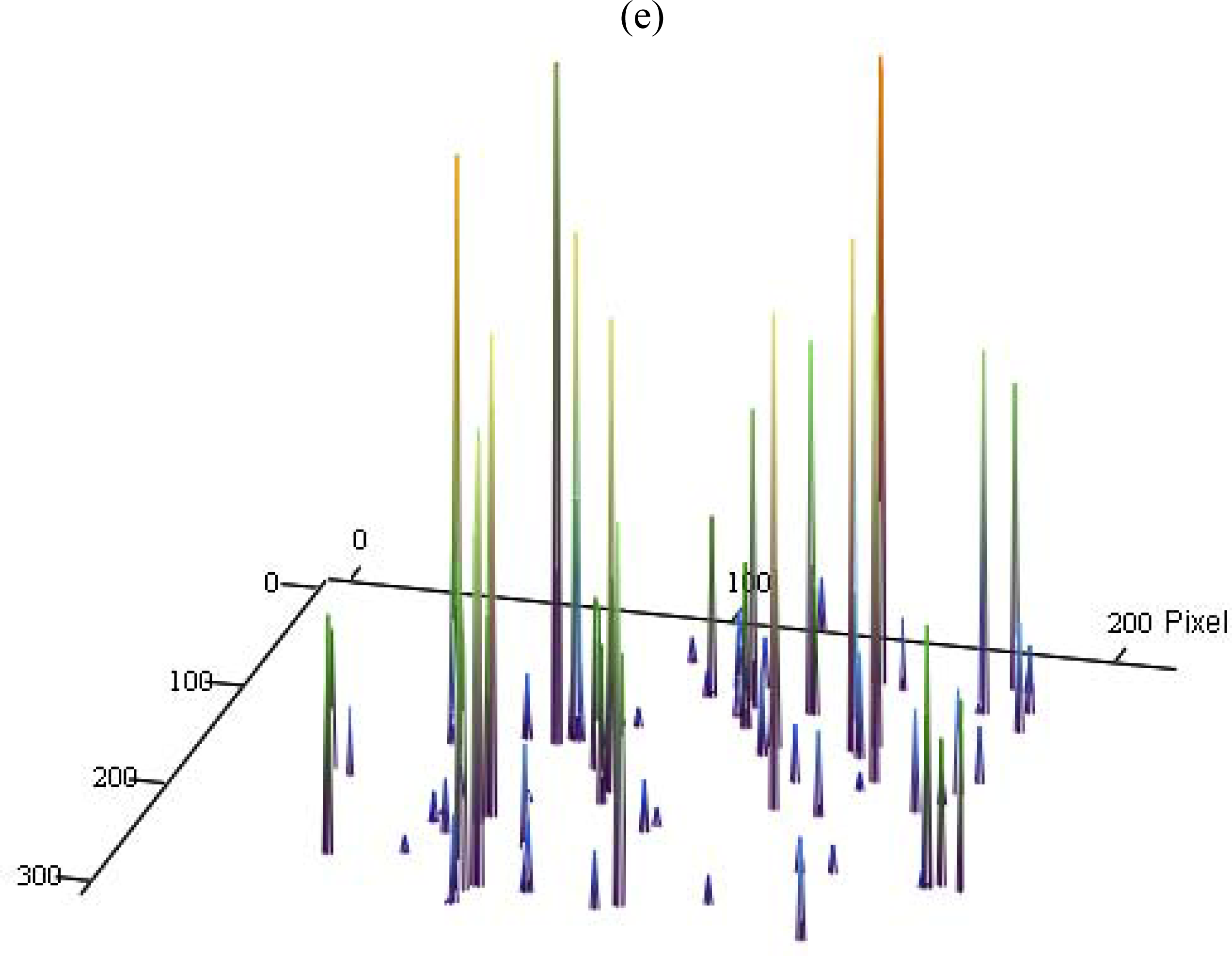
3.3. Single Molecule Detection
4. Discussion
Acknowledgments
References
- Pawley, J. Handbook of Biological Confocal Microscopy; Plenum Press: New York, NY, USA, 1990. [Google Scholar]
- Denk, W; Strickler, JH; Webb, WW. Two-photon laser scanning microscope. Science 1990, 248, 73–76. [Google Scholar]
- König, K. Multiphoton microscopy in life sciences. J. Microsc 2000, 200, 83–86. [Google Scholar]
- Willig, KI; Harke, B; Medda, R; Hell, SW. “STED microscopy with continuous wave beams”. Nat. Meth 2007, 4, 915–918. [Google Scholar]
- Coates, CG; Denvir, DJ; McHale, NG; Thornbury, KD; Hollywood, MA. Optimizing low-light microscopy with back-illuminated electron multiplying charge-coupled device: enhanced sensitivity, speed and resolution. J. Biomed. Opt 2004, 9, 1244–1252. [Google Scholar]
- Neil, MAA; Juskaitis, R; Wilson, T. Method of obtaining optical sectioning by structured light in a conventional microscope. Opt. Lett 1997, 22, 1905–1907. [Google Scholar]
- Huisken, J; Swoger, J; del Bene, F; Wittbrodt, J; Stelzer, EHK. Optical sectioning deep inside live embryos by SPIM. Science 2004, 305, 1007–1009. [Google Scholar]
- Betzig, E; Patterson, GH; Sougrat, R; Lindwasser, OW; Olenych, S; Bonifacino, JS; Davidson, MW; Lippincott-Schwartz, J; Hess, HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar]
- Rust, MJ; Bates, M; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Meth 2006, 3, 793–796. [Google Scholar]
- Hess, ST; Girirajan, TPK; Mason, MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J 2006, 91, 4258–4272. [Google Scholar]
- Schneckenburger, H. Total internal reflection fluorescence microscopy: Technical innovations and novel applications. Curr. Opin. Biotechnol 2005, 16, 13–18. [Google Scholar]
- Sako, Y; Uyemura, T. Total internal reflection fluorescence microscopy for single molecule imaging in living cells. Cell. Struct. Funct 2002, 27, 357–365. [Google Scholar]
- Parasassi, T; Krasnowska, EK; Bagatolli, L; Gratton, E. Laurdan and prodan as polaritysensitive fluorescent membrane probes. J. Fluoresc 1998, 4, 365–373. [Google Scholar]
- Stock, K; Sailer, R; Strauss, WSL; Lyttek, M; Steiner, R; Schneckenburger, H. Variable-angle total internal reflection fluorescence microscopy (VA-TIRFM): Realization and application of a compact illumination device. J. Microsc 2003, 211, 19–29. [Google Scholar]
- Wagner, M; Weber, P; Strauss, WSL; Lassalle, H-P; Schneckenburger, H. Nanotomography of cell surfaces with evanescent fields. Adv Opt Technol 2008, 2008, 254317:1–254317:7. [Google Scholar]
- Hockberger, P; Skimina, TA; Centonze, VE; Lavin, C; Chu, S; Dadras, S; Reddy, JK; White, JG. Activation of flavin-containing oxidases underlies light-induced production of H2O2 in mammalian cells. Proc. Natl. Sci. USA 1999, 96, 6255–6260. [Google Scholar]
- Vincent, SH. Oxidative effects of heme and porphyrins on proteins and lipids. Semin. Hematol 1989, 26, 105–113. [Google Scholar]
- Ricchelli, F. Photophysical properties of porphyrins in biological membranes. J. Photochem. Photobiol. B: Biol 1995, 29, 109–118. [Google Scholar]
- Dougherty, TJ; Marcus, SL. Photodynamic therapy. Eur. J. Cancer 1992, 28A, 1734–1742. [Google Scholar]
- Schneckenburger, H; Weber, P; Wagner, M; Schickinger, S; Bruns, T; Strauss, WSL. Dose limited fluorescence microscopy of 5-aminolevulinic acid induced protoporphyrin IX in living cells? Proc. SPIE 2009, 10, 11640F:1–71640F:7. [Google Scholar]
- Hoebe, RA; van Oven, CH; Gadella, TWJ, Jr; Dhonukshe, PB; van Noorden, CJF; Manders, EMM. Controlled light-exposure microscopy reduces photobleaching and phototoxicity in fluorescence live-cell imaging. Nat. Biotechnol 2007, 25, 249–253. [Google Scholar]
- Murray, JM; Appleton, PL; Swedlow, JR; Waters, JC. Evaluating performance in three-dimensional fluorescence microscopy. J. Microsc 2007, 228, 390–405. [Google Scholar]




© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wagner, M.; Weber, P.; Bruns, T.; Strauss, W.S.L.; Wittig, R.; Schneckenburger, H. Light Dose is a Limiting Factor to Maintain Cell Viability in Fluorescence Microscopy and Single Molecule Detection. Int. J. Mol. Sci. 2010, 11, 956-966. https://doi.org/10.3390/ijms11030956
Wagner M, Weber P, Bruns T, Strauss WSL, Wittig R, Schneckenburger H. Light Dose is a Limiting Factor to Maintain Cell Viability in Fluorescence Microscopy and Single Molecule Detection. International Journal of Molecular Sciences. 2010; 11(3):956-966. https://doi.org/10.3390/ijms11030956
Chicago/Turabian StyleWagner, Michael, Petra Weber, Thomas Bruns, Wolfgang S. L. Strauss, Rainer Wittig, and Herbert Schneckenburger. 2010. "Light Dose is a Limiting Factor to Maintain Cell Viability in Fluorescence Microscopy and Single Molecule Detection" International Journal of Molecular Sciences 11, no. 3: 956-966. https://doi.org/10.3390/ijms11030956