Molecular Characteristics of Kraft-AQ Pulping Lignin Fractionated by Sequential Organic Solvent Extraction

Abstract
:
1. Introduction
2. Results and Discussion
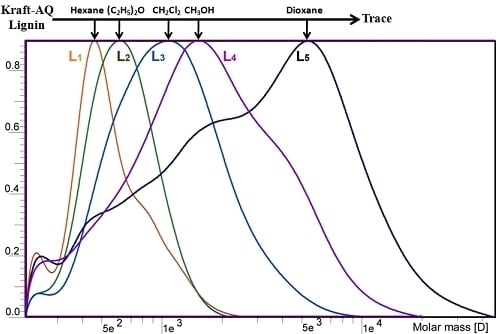
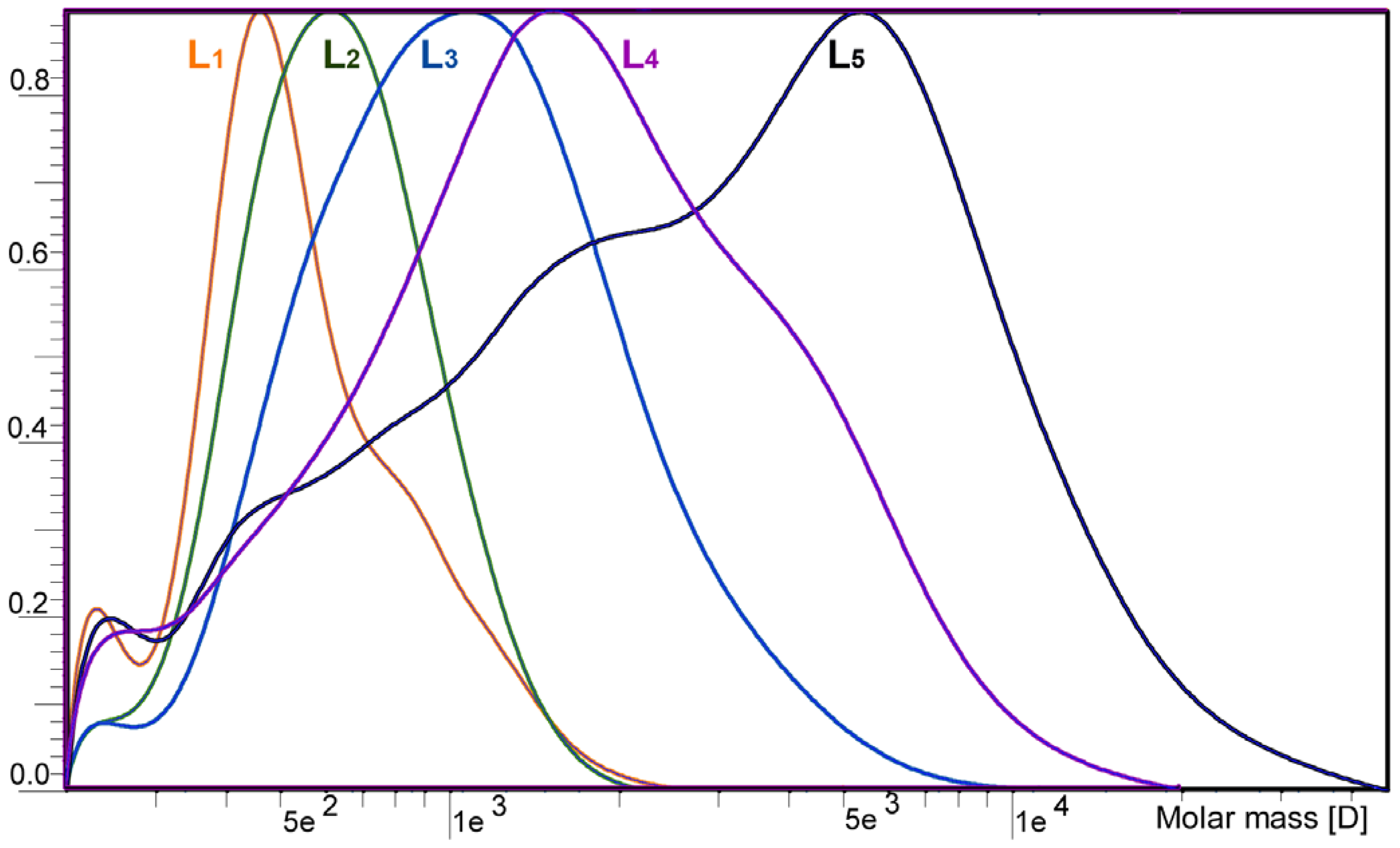
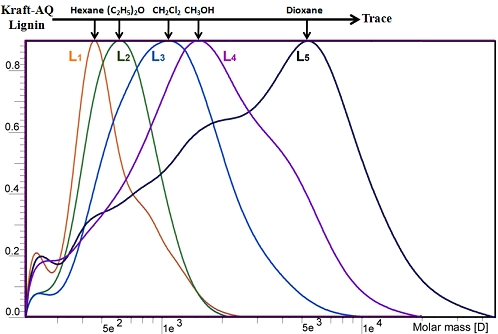
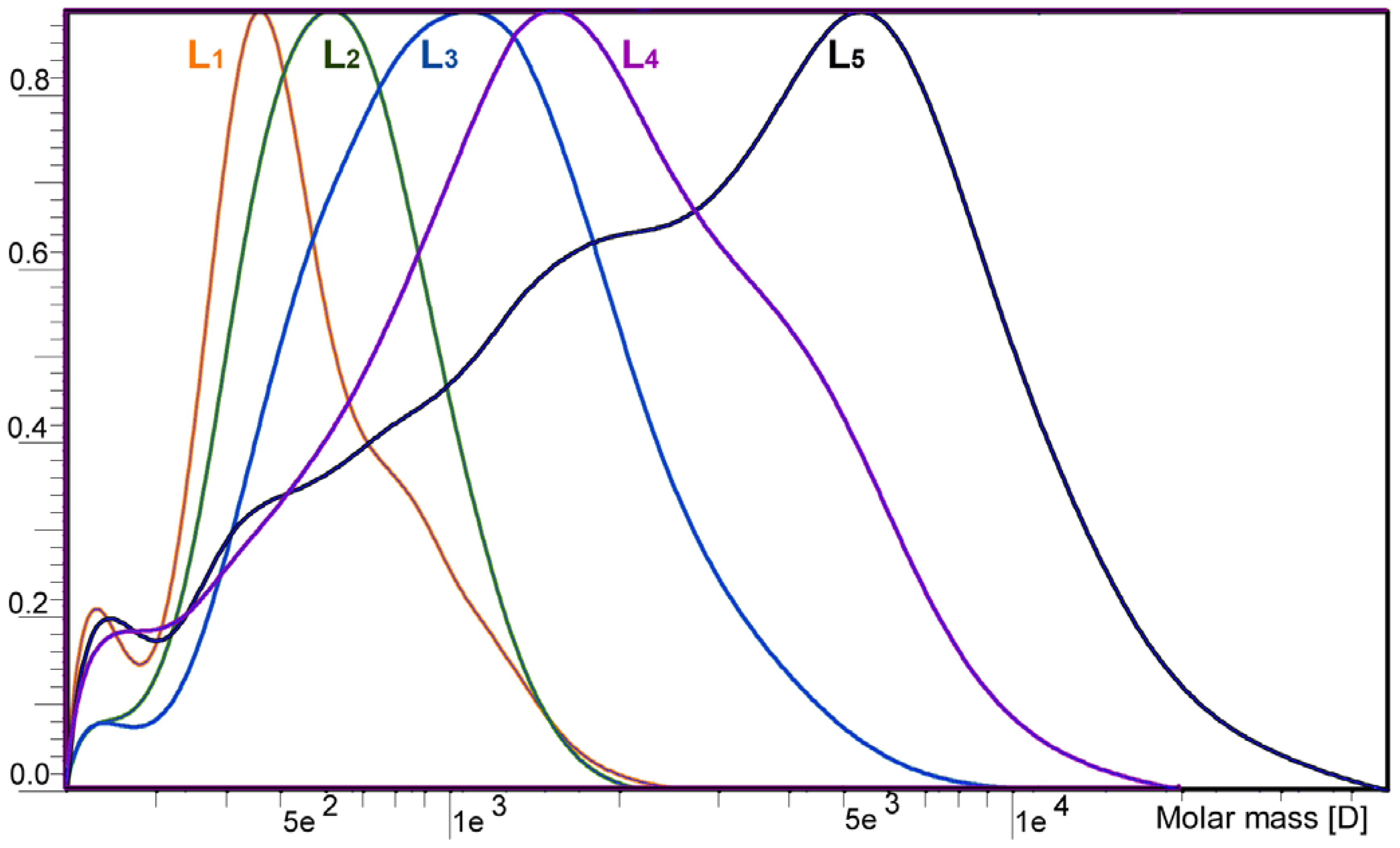
2.1. Yield and Molecular Weight Distribution
2.2. Composition of Phenolic Acids and Aldehydes
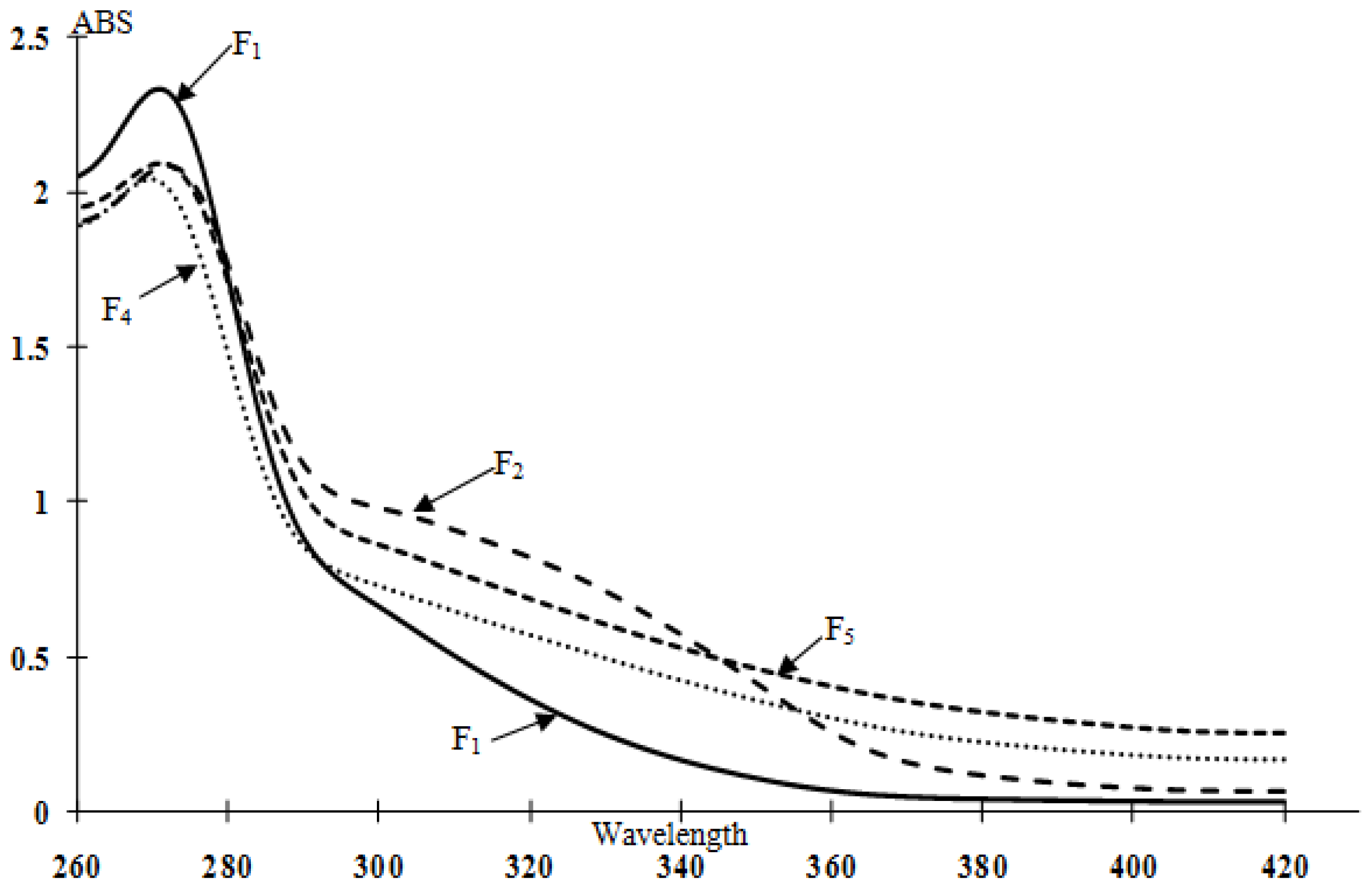
2.3. UV Analysis
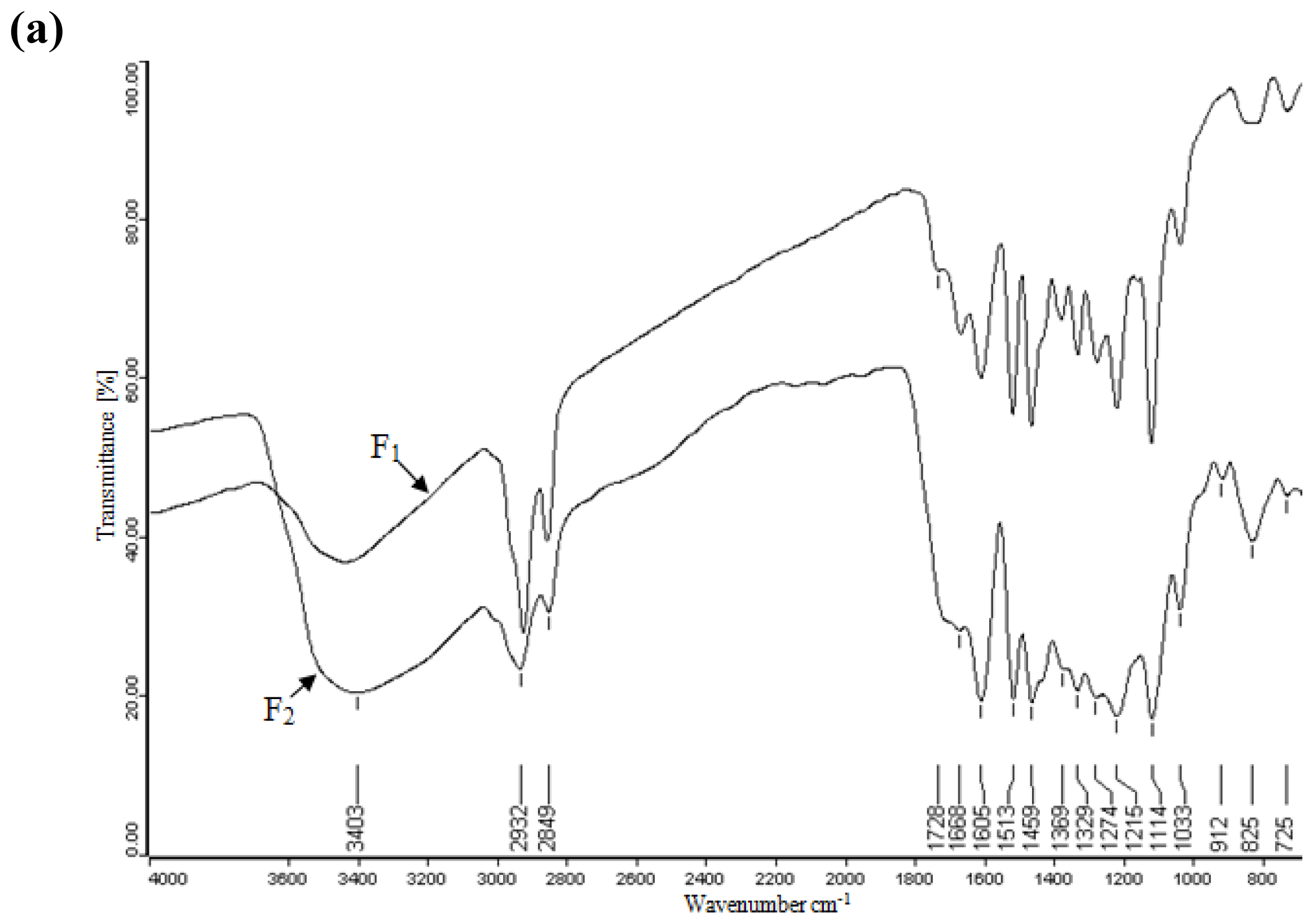
2.4. FT-IR Spectra Analysis
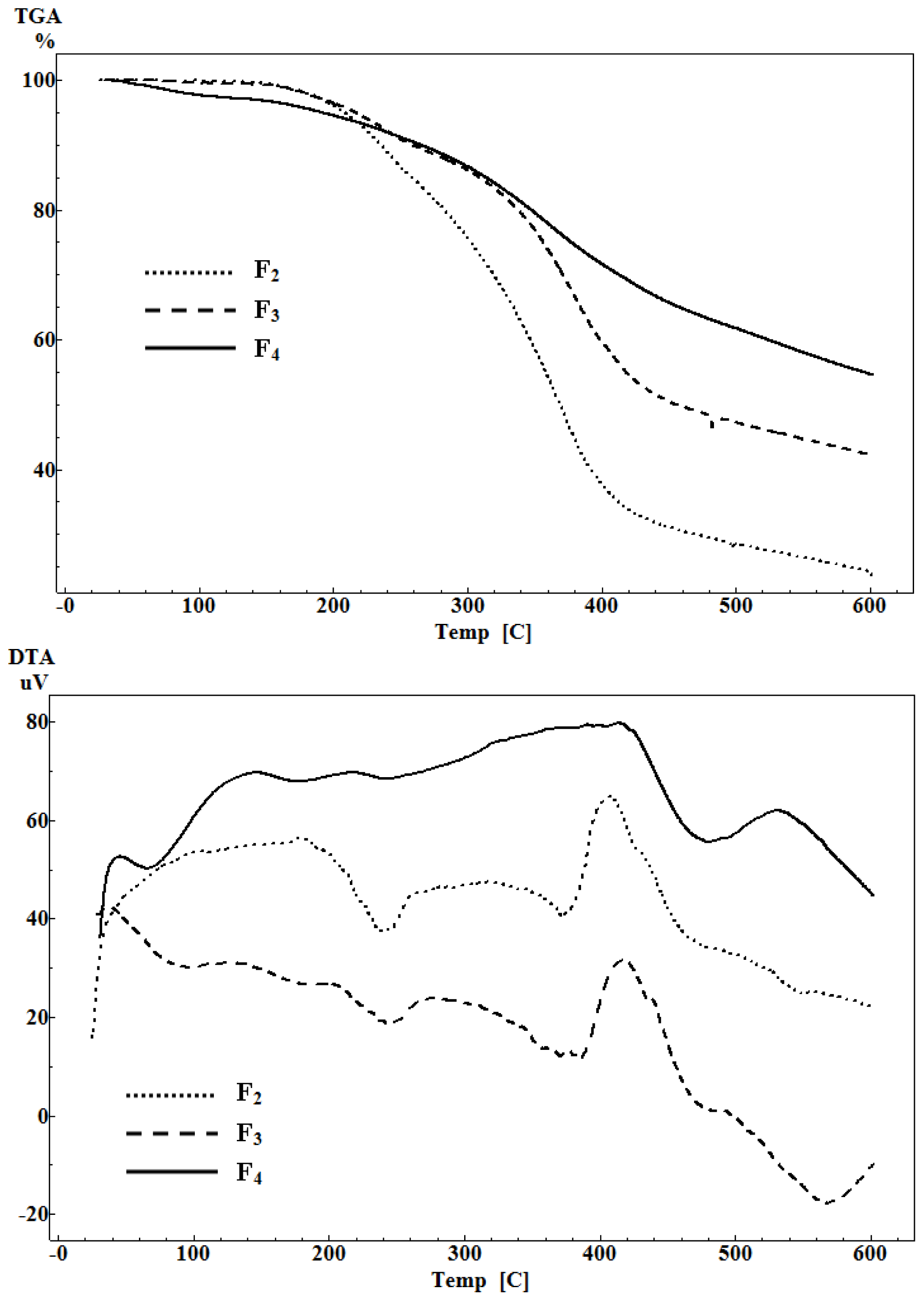
2.5. Thermal Analysis
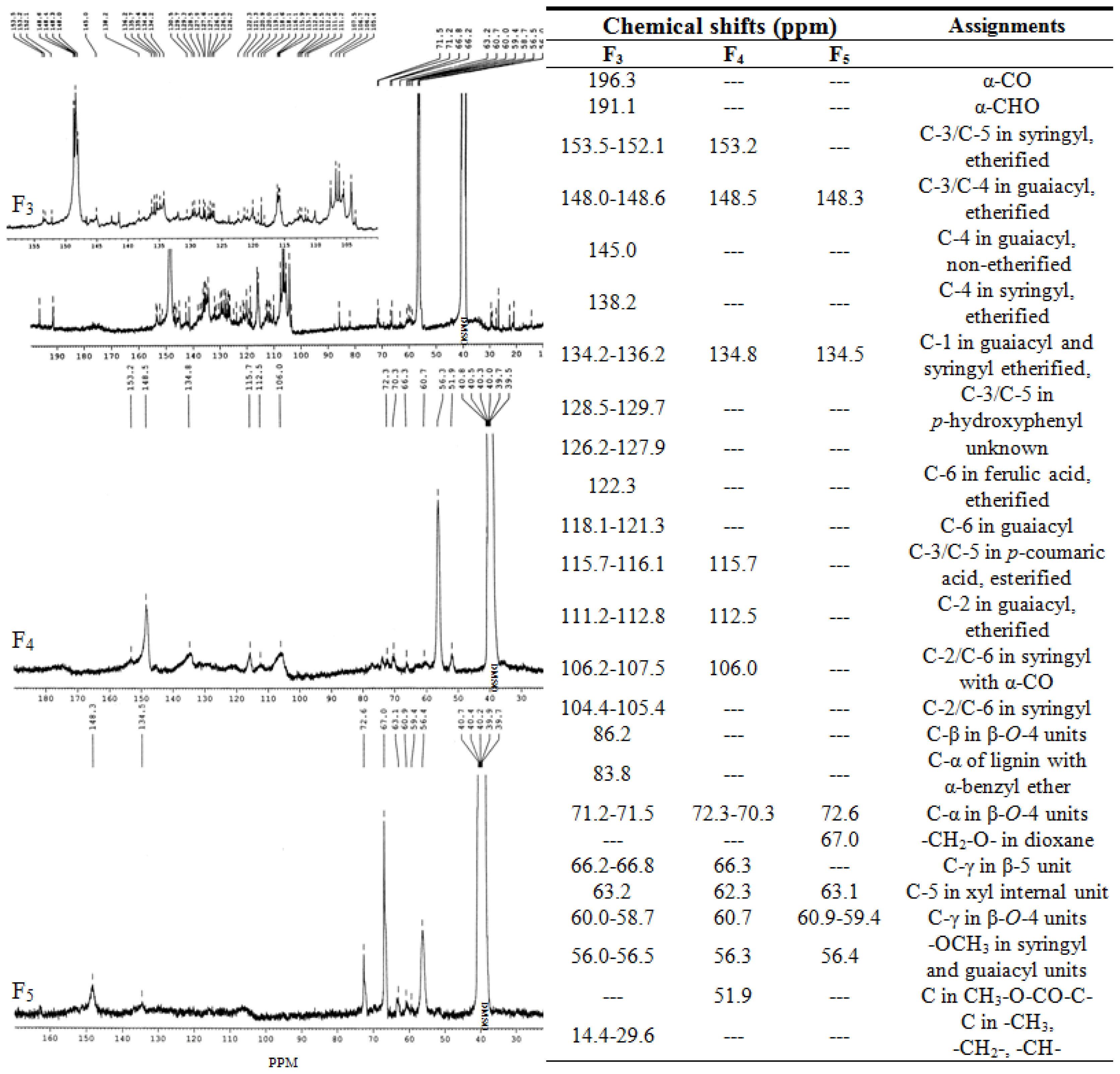
2.6. 13C NMR Spectra Analysis
3. Experimental Section
3.1. Kraft-AQ Pulping
3.2. Lignin Recovery from Black Liquor
3.3. Separation Process
3.4. Analytical Method
4. Conclusions
Acknowledgements
References and Notes
- Chow, J; Kopp, RJ; Portney, PR. Energy resources and global development. Science 2003, 302, 1528–1531. [Google Scholar]
- Kocurek, MJ; Grace, TM; Malcolm, E. Alkaline pulping. In Pulp and Paper Manufacture Series, 3rd ed; Tappi Press: Atlanta, GA, USA, 1989; Volume 5, pp. 248–273. [Google Scholar]
- Fleming, BI; Kubes, GJ; MacLeod, JM; Bolker, HI. Soda pulping with anthraquinone. Tappi J 1978, 61, 43–46. [Google Scholar]
- Lowendahl, L; Samuelson, O. Carbohydrate stabilization during soda pulping with addition of anthraquinone. Tappi J 1978, 61, 19–21. [Google Scholar]
- Vuorinen, T. The role of carbohydrates in alkali anthraquinone pulping. J. Wood Chem. Technol 1993, 13, 97–125. [Google Scholar]
- Allen, VN; Hatton, JV; Gee, WY. Effect of sulfidity in alkaline pulping of white spruce with anthraquinone. Tappi J 1981, 64, 64–66. [Google Scholar]
- Mohan, VS; Karthikeyan, J. Removal of lignin and tannin colour from aqueous solution by adsorption onto activated charcoal. Environ. Pollut 1997, 97, 183–187. [Google Scholar]
- Zhang, QL; Chuang, KT. Adsorption of organic pollutants from effluents of a Kraft pulp mill on activated carbon and polymer resin. Adv. Environ. Res 2001, 5, 251–258. [Google Scholar]
- Braun, JL; Holtman, KM; Kadla, JF. Lignin-based carbon fibers: oxidative thermostabilization of kraft lignin. Carbon 2005, 43, 384–394. [Google Scholar]
- Kadla, JF; Kubo, S; Venditti, RA; Gilbert, RD; Compere, AL; Griffith, W. Lignin-based carbon fibers for composite fiber applications. Carbon 2002, 40, 2913–2920. [Google Scholar]
- Vazquez, G; Rodriguez-Bona, C; Freire, S; Gonzalez-Alvarez, J; Antorrena, G. Acetosolv pine lignin as copolymer in resins for manufacture of exterior grade plywoods. Bioresour. Technol 1999, 70, 209–214. [Google Scholar]
- Hatakeyama, H; Kosugi, R; Hatakeyama, T. Thermal properties of lignin-and molasses-based polyurethane foams. J. Therm. Anal. Calorim 2008, 92, 419–424. [Google Scholar]
- Lora, JH; Glasser, WG. Recent industrial applications of lignin: A sustainable alternative to nonrenewable materials. J. Polym. Environ 2002, 10, 39–48. [Google Scholar]
- Stewart, D. Lignin as a base material for materials applications: Chemistry, application and economics. Ind. Crop. Prod 2008, 27, 202–207. [Google Scholar]
- Mörck, R; Reimann, A; Kringstad, KP. Fractionation of kraft lignin by successive extraction with organic solvents. Ø. Fractionation of kraft lignin from birch. Holzforschung 1988, 42, 111–116. [Google Scholar]
- Mörck, R; Yoshida, H; Kringstad, KP. Fractionation of kraft lignin by successive extraction with organic solvents. ². Functional groups, 13C-NMR-spectra and molecular weight distributions. Holzforschung 1986, 40, 51–60. [Google Scholar]
- van der Klashorst, GH; Strauss, HF. Properties and potential utilization of industrial eucalyptus soda/anthraquinone lignin. Part ². Isolation, identification and origin of the low molecular mass lignin fragments present in an industrial soda/anthraquinone spent pulping liquor. Holzforschung 1987, 41, 123–131. [Google Scholar]
- Thring, RW; Vanderlaan, MN; Griffin, SL. Fractionation of ALCELL® lignin by sequential solvent extraction. J. Wood Chem. Technol 1996, 16, 139–154. [Google Scholar]
- Sun, RC; Lu, Q; Sun, XF. Physico-chemical and thermal characterization of lignins from Caligonum monogoliacum and Tamarix spp. Polym. Degrad. Stab 2001, 72, 229–238. [Google Scholar]
- Xu, F; Sun, RC; Zhai, MZ; Sun, JX; Jiang, JX; Zhao, GJ. Comparative study of three lignin fractions isolated from mild ball-milled Tamarix austromogoliac and Caragana sepium. J. Appl. Polym. Sci 2008, 108, 1158–1168. [Google Scholar]
- Scalbert, A; Monties, B; Guittet, E; Lallemand, JY. Comparison of wheat straw lignin preparation. I. Chemical and spectroscopic characterization. Holzforschung 1986, 40, 119–127. [Google Scholar]
- Sun, XF; Xu, F; Sun, RC; Fowler, P; Baird, MS. Characteristics of degraded cellulose obtained from steam-exploded wheat straw. Carbohydr. Res 2005, 340, 97–106. [Google Scholar]
- Balat, M. Mechanism of thermochemical biomass conversion processes. Part 1: Reactions of Pyrolysis. Energ. Resour. Part A 1981, 30, 620–635. [Google Scholar]
- Oren, MJ; Nassar, MM; Mackay, GDM. Infrared study of inert carbonization of spruce wood lignin under helium atmosphere. Can. J. Spectrosc 1984, 29, 10–12. [Google Scholar]
- Fenner, RA; Lephardt, JO. Examination of the thermal decomposition of kraft pine lignin by Fourier transform infrared evolved gas analysis. J. Agric. Food Chem 1981, 29, 846–849. [Google Scholar]
- Gardner, DJ; Schultz, TP; McGinnis, GD. The pyrolytic behavior of selected lignin preparations. J. Wood Chem. Tech 1985, 5, 85–110. [Google Scholar]
- Sun, RC; Tomkinson, J; Jones, GL. Fractional characterization of ash-AQ lignin by successive extraction with organic solvents from oil palm EFB fiber. Polym. Degrad. Stab 2000, 68, 111–119. [Google Scholar]
- Holtman, KM; Chang, HM; Jameel, H. Quantitative 13-C NMR characterization of milled wood lignins isolated by different milling techniques. J. Wood Chem. Technol 2006, 26, 21–34. [Google Scholar]
- Lapierre, C; Monties, B; Guittet, E; Lallemand, JY. The quantitative measurements in hardwood lignin 13C NMR spectra. Holzforschung 1985, 39, 367–368. [Google Scholar]
- Xie, YM; Terashima, N. Selective carbon 13-enrichment of side chain carbons of rice stalk lignin traced by carbon 13 nuclear magnetic resonance. Mokuzai Gakkaishi 1993, 39, 91–97. [Google Scholar]
- Sun, RC; Tomkinson, J; Mao, FC; Sun, XF. Physicochemical characterization of lignins from rice straw by hydrogen peroxide treatment. J. Appl. Polym. Sci 2001, 79, 719–732. [Google Scholar]
- Kim, H; Hill, MK; Fricke, AL. Preparation of kraft lignin from black liquor. Tappi J 1987, 11, 112–116. [Google Scholar]
- Schuerch, C. The solvent properties of liquids and their relation to the solubility, swelling, isolation and fractionation of lignin. J. Am. Chem. Soc 1952, 74, 5061–5067. [Google Scholar]
- Pellinen, J; Salkinoja-Salonen, M. High-performance size-exclusion chromatography of lignin and its derivatives. J. Chromatogr 1985, 328, 299–308. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin fractionsa | |||||
|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | |
| Yieldbc | 0.2 | 1.6 | 33.4 | 59.1 | 4.4 |
| —,Mw | 493 | 686 | 1174 | 2468 | 13651 |
| —,Mn | 492 | 665 | 1012 | 1675 | 3053 |
| —,Mw/—,Mn | 1.0 | 1.0 | 1.2 | 1.5 | 4.5 |
| Phenolic acids and aldehydes | Lignin fractionsa | ||||
|---|---|---|---|---|---|
| F1 | F2 | F3 | F4 | F5 | |
| Vanillic acid | 5.7 | 6.5 | 7.8 | 5.4 | 9.1 |
| Vanillin | 21.1 | 23.1 | 23.8 | 24.9 | 25.9 |
| Acetovanillone | 6.0 | 6.2 | 8.2 | 9.3 | 6.3 |
| Syringic acid | 2.9 | 10.4 | 11.6 | 15.8 | 22.9 |
| Syringaldehyde | 28.9 | 29.1 | 30.1 | 28.2 | 25.3 |
| Acetosyringone | 20.0 | 16.6 | 16.5 | 13.2 | 9.8 |
| p-Hydroxybezonic acid | 15.0 | 7.6 | 1.7 | 2.8 | NDb |
| p-Hydroxybenzaldehyde | 0.4 | 0.5 | 0.3 | 0.4 | 0.7 |
| S/Gc | 1.58 | 1.57 | 1.46 | 1.44 | 1.40 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, K.; Xu, F.; Sun, R. Molecular Characteristics of Kraft-AQ Pulping Lignin Fractionated by Sequential Organic Solvent Extraction. Int. J. Mol. Sci. 2010, 11, 2988-3001. https://doi.org/10.3390/ijms11082988
Wang K, Xu F, Sun R. Molecular Characteristics of Kraft-AQ Pulping Lignin Fractionated by Sequential Organic Solvent Extraction. International Journal of Molecular Sciences. 2010; 11(8):2988-3001. https://doi.org/10.3390/ijms11082988
Chicago/Turabian StyleWang, Kun, Feng Xu, and Runcang Sun. 2010. "Molecular Characteristics of Kraft-AQ Pulping Lignin Fractionated by Sequential Organic Solvent Extraction" International Journal of Molecular Sciences 11, no. 8: 2988-3001. https://doi.org/10.3390/ijms11082988