Quantitative Analysis of Cellular Metabolic Dissipative, Self-Organized Structures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction to Molecular Self-Organization in the Cellular Metabolism
1.1. Supramolecular Self-Organization of the Catalytic Activities
1.2. Structural Microcompartmentalization of the Metabolic Processes
1.3. Metabolic Temporal Self-Organizations
1.4. Metabolic Temporal Self-Organizations with a Period of 24 Hours
1.5. Metabolic Temporal-Spatial Self-Organizations
1.6. Global Self-Organized Metabolic Structures
1.7. Quantitative Analysis of Functional Metabolic Structures
2. Dissipative Structures: Thermodynamic Aspects of Self-Organization
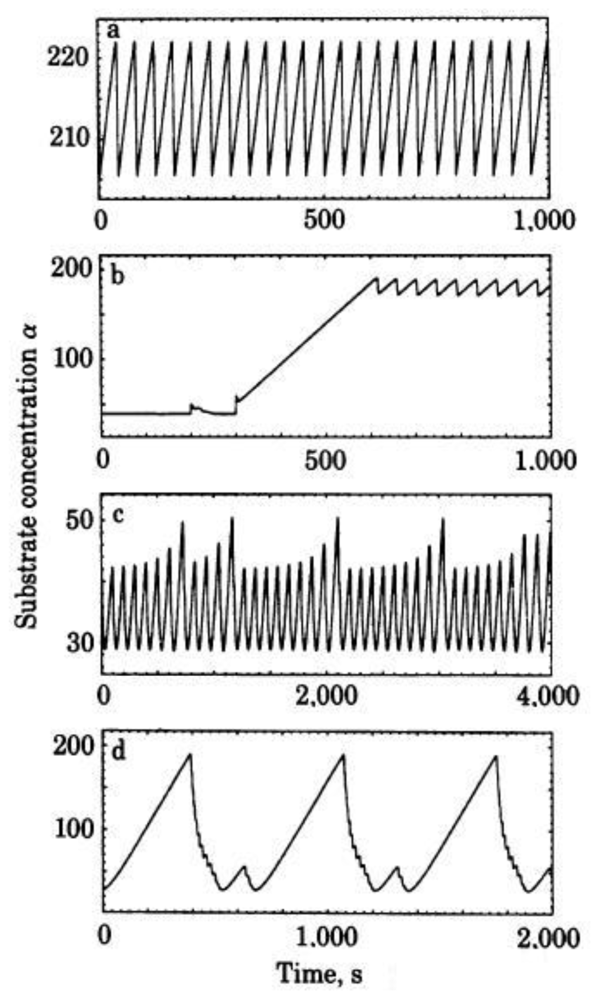
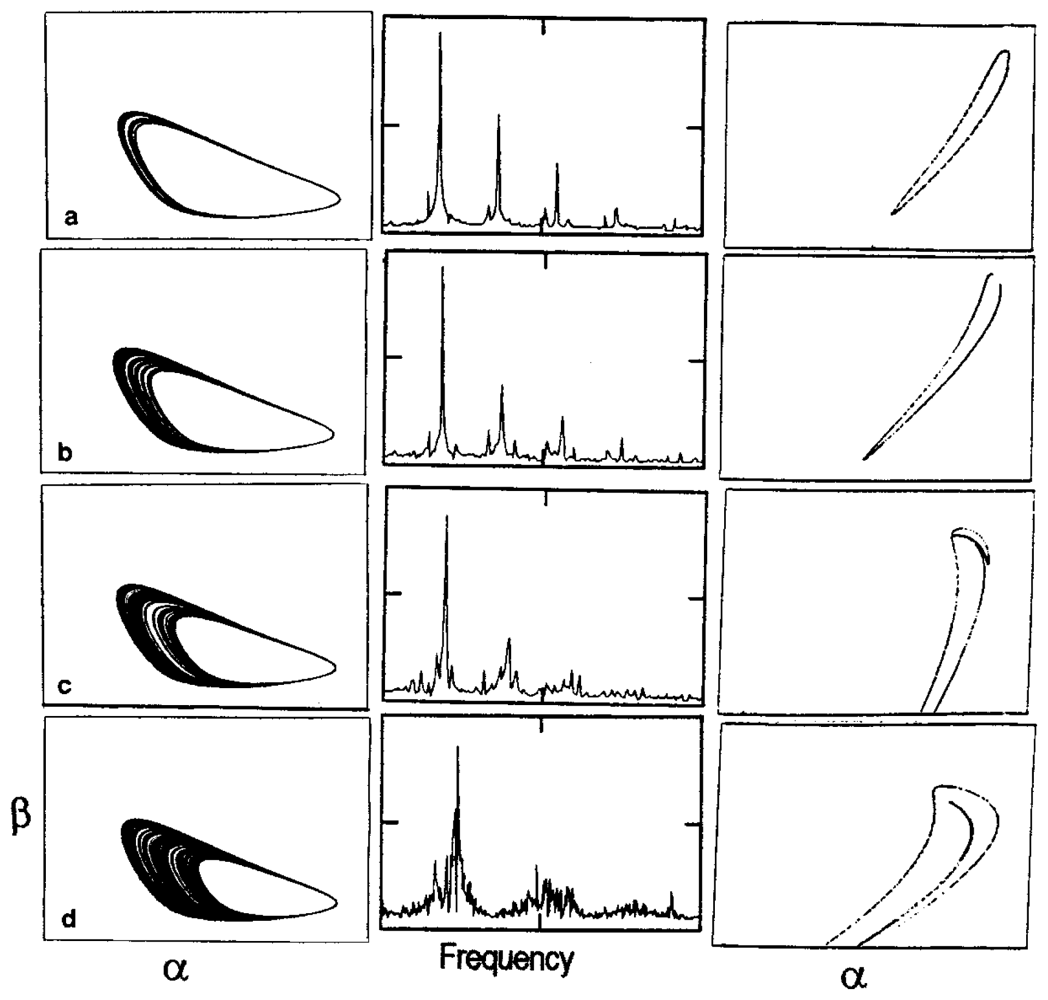
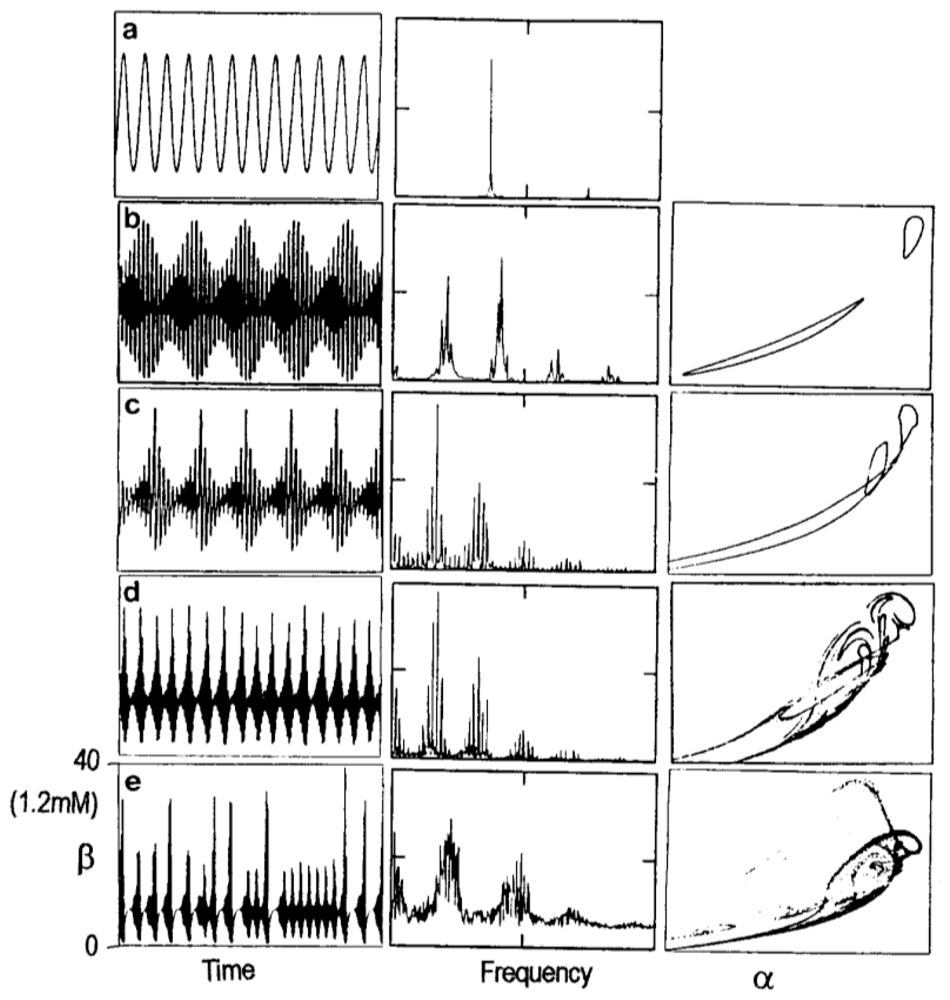
3. Dissipative Self-Organization and Temporal Metabolic Patterns
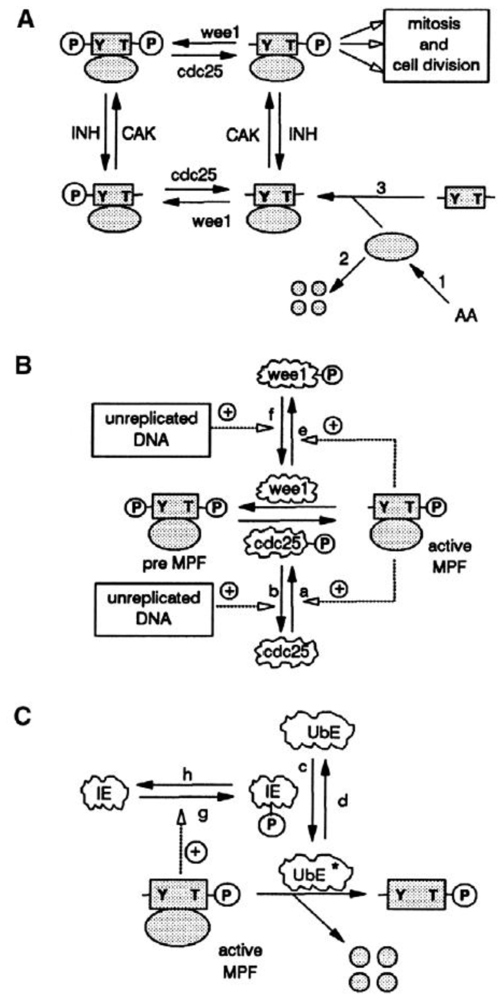
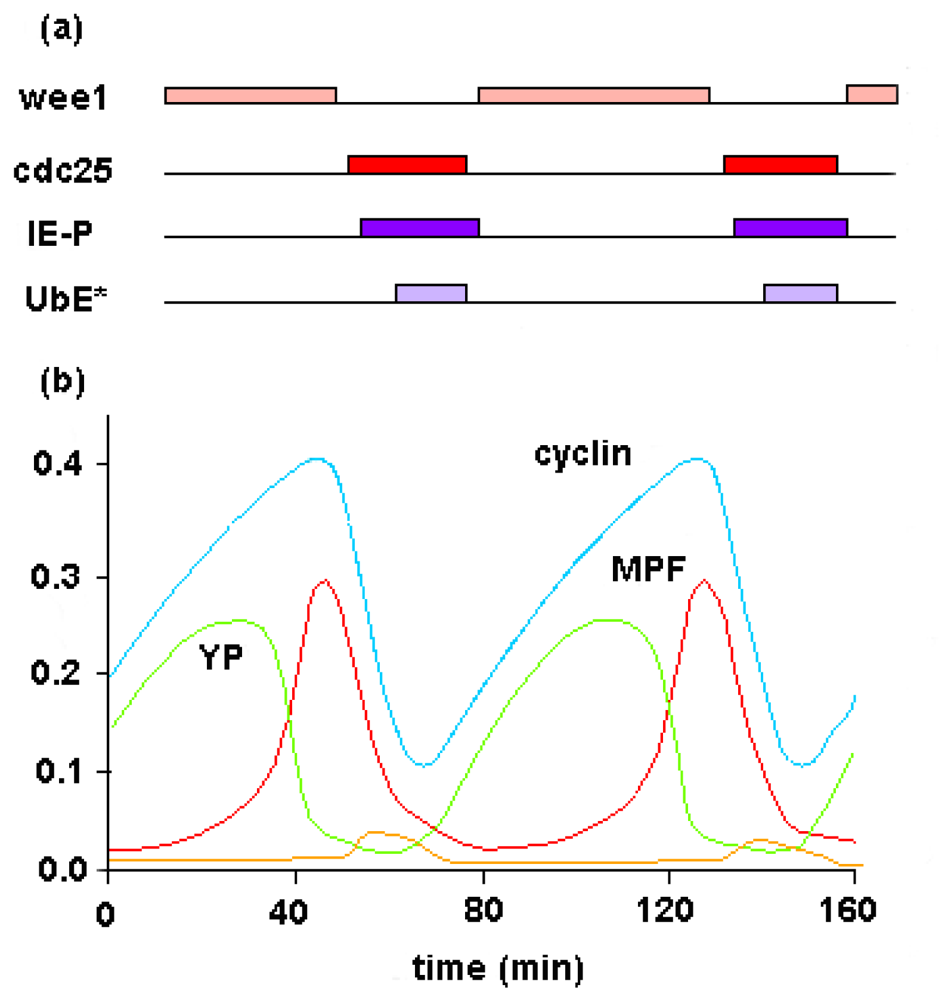
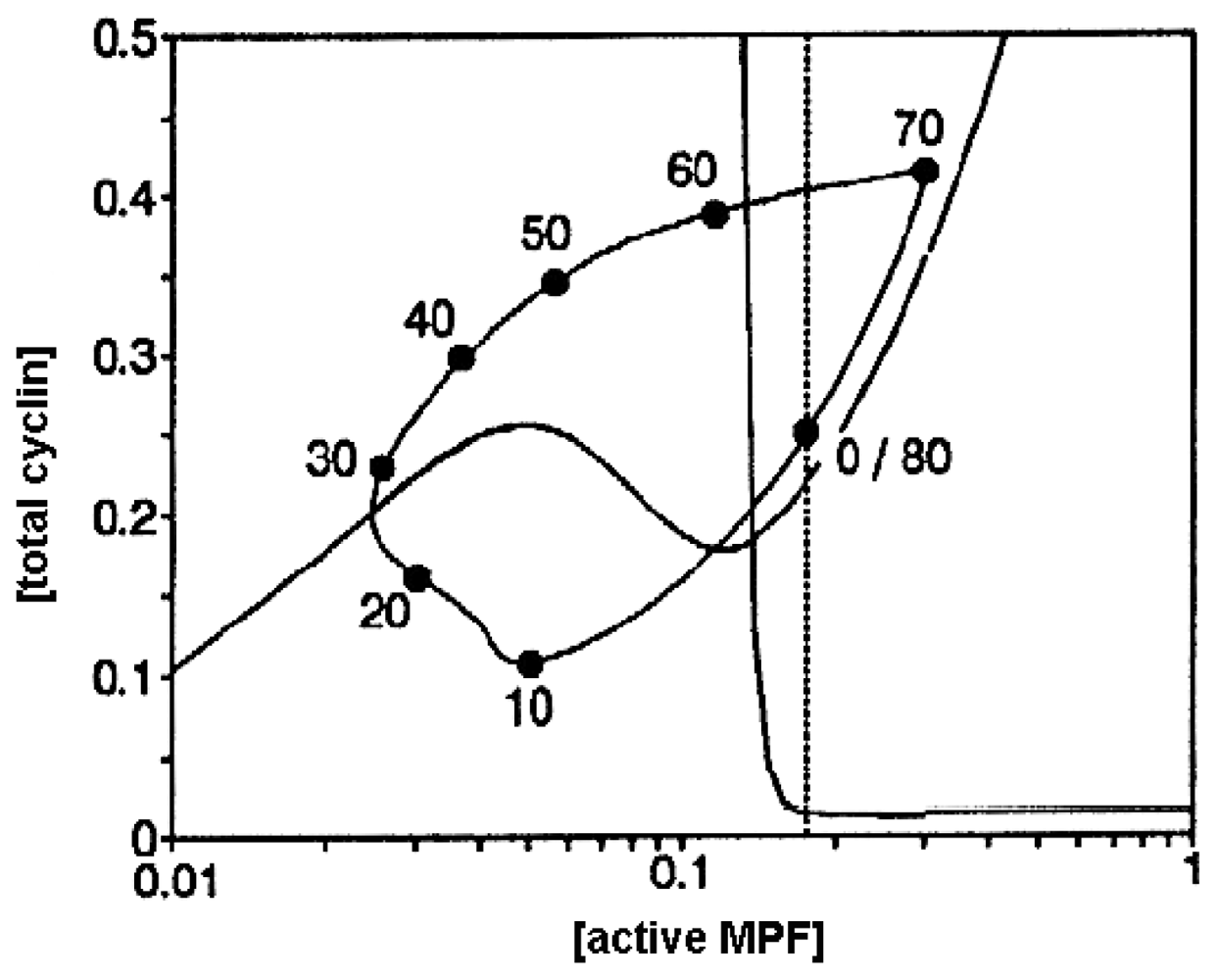
4. Metabolic Self-Organization and the Cell-Cycle
5. Quantitative Analysis in Metabolic Networks
6. Effect of the Delays on Temporal Self-Organizations
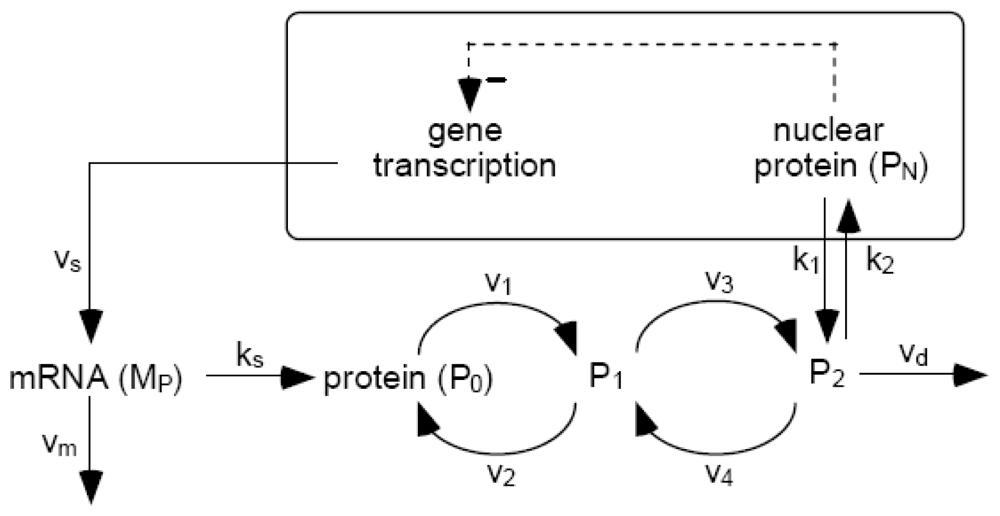
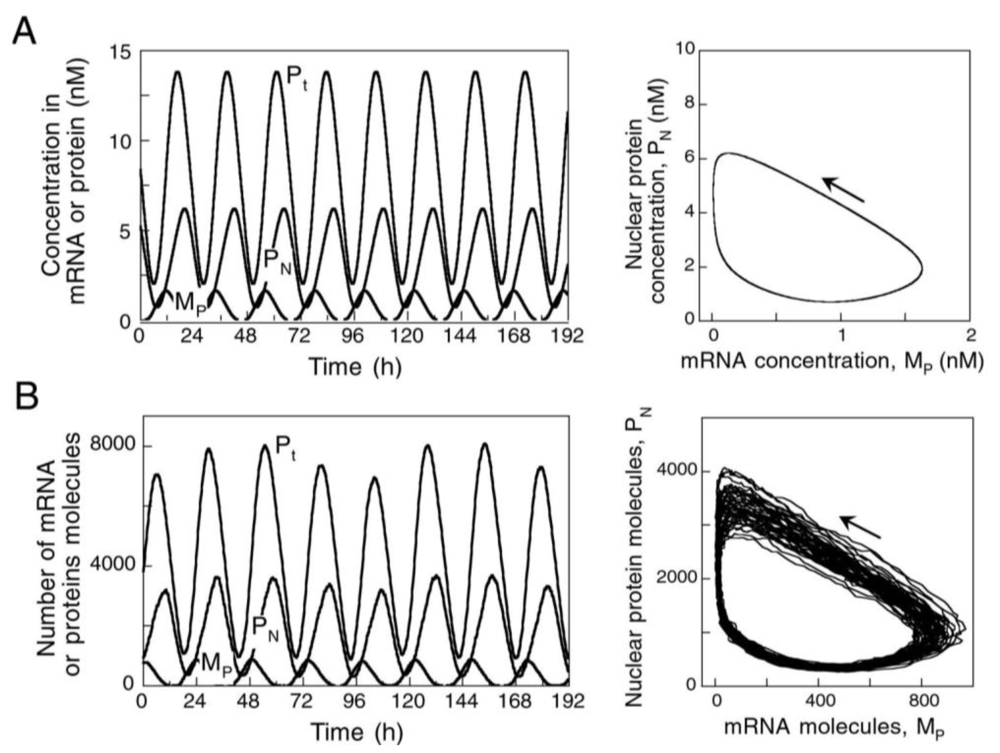
7. Self-Organizations in Stochastic Processes: Genetic Expression
8. Metabolic Attractors
- It is impossible to go out; in other words, if y(t0) is in A for some time t0, later y(t) remains in A.
- There exists a neighbourhood of itself B (basin of attraction) such that for any initial condition in B, the system approaches A indefinitely.
- A is a compact set; this means it is a closed and bounded set.
9. Stability in Dynamical Behaviors: The Maximal Lyapunov Exponent
10. Long-Term Correlations in Metabolic Activities
- Partition of the data points in adjacent non-overlapping windows of size n, where Wi = {x(i−1)n+1,..., xin}. If N is not a power of 2 and N is not divisible by n, then the last remaining points are ignored for this value of n. For instance, if N = 31 and n = 4, the first 28 points are partitioned into seven windows.
- Subtraction of the line between the first and last points for the points in the n-th window.
- For each , calculation of the standard deviation SDi of the points in each window, by using the formula:
- where x̄i is the average in the window Wi.
- Evaluation of the average of the standard deviations corresponding to Equation (5).
- Observation of the range of the window sizes n over which the regression line of versus log (n) gives a good fit (usually some initial and end pairs are excluded).
- In this range, the slope of the regression line gives the estimation of the Hurst coefficient H.
11. Measure of Complexity. Kolmogorv-Sinai Entropy
12. Conclusions
References
- Jeong, H; Tombor, B; Albert, R; Oltvai, ZN; Barabási, AL. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar]
- Sear, RP. The cytoplasm of living cells: a functional mixture of thousands of components. J. Phys. Condens. Matter 2005, 17, S3587–S3595. [Google Scholar]
- Goldbeter, A. Biological rhythms as temporal dissipative structures. Adv. Chem. Phys 2007, 135, 253–295. [Google Scholar]
- Nicolis, G; Prigogine, I. Self-organization in nonequilibrium systems. In From dissipative structures to order through fluctuations; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Gavin, AC; Bosche, M; Krause, R; Grandi, P; Marzioch, M; Bauer, A; Schultz, J; Rick, JM; Michon, AM; Cruciat, CM; Remor, M; Höfert, C; Schelder, M; Brajenovic, M; Ruffner, H; Merino, A; Klein, K; Hudak, M; Dickson, D; Rudi, T; Gnau, V; Bauch, A; Bastuck, S; Huhse, B; Leutwein, C; Heurtier, MA; Copley, R; Edelmann, A; Querfurth, E; Rybin, V; Drewes, G; Raida, M; Bouwmeester, T; Bork, P; Seraphin, B; Kuster, B; Neubauer, G; Superti-Furga, G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 2002, 415, 141–147. [Google Scholar]
- Fields, S; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar]
- de Graaf, RA; Van Kranenburg, A; Nicolay, K. In vivo 31P-NMR spectroscopy of ATP and phosphocreatine in rat skeletal muscle. Biophys. J 2000, 78, 1657–1664. [Google Scholar]
- Ho, Y; Gruhler, A; Heilbut, A; Bader, DG; Moore, L; Adams, SL; Millar, A; Taylor, P; Bennett, K; Boutilier, K; Yang, L; Wolting, Ch; Donaldson, I; Schandorff, S; Shewnarane, J; Vo, M; Taggart, J; Goudreaul, M; Muskat, B; Alfarano, C; Dewar, D; Lin, Z; Michalickova, K; Willems, AR; Sassi, H; Nielsen, PA; Rasmussen, KJ; Andersen, JR; Johansen, LE; Hansen, LH; Jespersen, H; Podtelejnikov, A; Nielsen, E; Crawford, J; Poulsen, V; S⊘rensen, BD; Matthiesen, J; Hendrickson, RC; Gleeson, F; Pawson, T; Moran, MF; Durocher, D; Mann, M; Hogue, CWV; Figeys, D; Tyers, M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180–183. [Google Scholar] [Green Version]
- Ito, T; Chiba, T; Ozawa, R; Yoshida, M; Hattori, M; Sakaki, Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 2001, 98, 4569–4574. [Google Scholar]
- Uetz, P; Giot, L; Cagney, G; Mansfield, TA; Judson, RS; Knight, JR; Lockshon, D; Narayan, V; Srinivasan, M; Poschart, P; Qureshi-Emili, A; Li, Y; Godwin, B; Conover, D; Kalbfleisch, T; Vijayadamodar, G; Yang, M; Johnston, M; Fields, S; Rothberg, JM. A comprehensive analysis of proteinprotein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar]
- Mowbray, J; Moses, V. The tentative identification in Escherichia coli of a multienzyme complex with glycolytic activity. J. Biochem 1976, 66, 25–36. [Google Scholar]
- Barnes, SJ; Weitzman, DP. Organization of citric acid cycle enzymes into a multienzyme cluster. FEBS Lett 1986, 201, 267–270. [Google Scholar]
- Porpaczy, Z; Sumegi, B; Alkonyi, I. Integration between NAD-dependent isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase complex, and NADH: Ubiquinone oxidoreductase. J. Biol. Chem 1987, 262, 9509–9514. [Google Scholar]
- Beeckmans, S; Kanarek, L. Demostration of physical interactions between consecutive enzymes of citric acid cycle and aspartate-malate shuttle. Eur. J. Biochem 1981, 117, 527–535. [Google Scholar]
- Kholodenko, BN; Lyubarev, AE; Kurganov, BI. Control of the metabolic flux in the system with high enzyme concentrations and moiety-conserved cycles: The sum of the flux control coefficients can drop significantly below unity. Eur. J. Biochem 1992, 210, 147–153. [Google Scholar]
- Kurganov, BI. The concept of biochemical organization. Trends Biochem. Sci 1993, 18, 405–406. [Google Scholar]
- Srere, PA; Sherry, AD; Malloy, CR; Sumegi, B. Channeling in Intermediary Metabolism; Portland Press Ltd: London, UK, 1997. [Google Scholar]
- Lyubarev, AE; Kurganov, BI. Origin of biochemical organization. BioSystems 1997, 42, 103–110. [Google Scholar]
- Welch, GR. On the role of organized multienzyme systems in cellular metabolism: A general synthesis. Prog. Biophys. Mol. Biol 1977, 32, 103–191. [Google Scholar]
- Welch, GR. Organized Multienzyme Systems; Academic Press: Orlando, FL, USA, 1985. [Google Scholar]
- Clegg, JS; Jackson, SA. Glucose metabolism and the channelling of glycolytic intermediates in permeabilized L-929 cells. Arch. Biochem. Biophys 1990, 278, 452–460. [Google Scholar]
- Negrutskii, BS; Deutscher, MP. Channeling of aminoacyl tRNA for protein synthesis in vivo. Proc. Natl. Acad. Sci. USA 1991, 88, 4991–4995. [Google Scholar]
- Ovadi, J; Srere, PA. Macromolecular compartmentation and channeling. Int. Rev. Cytol 2000, 192, 255–280. [Google Scholar]
- Jorgensen, K; Rasmussen, AV; Morant, M; Nielsen, AH; Bjarnholt, N; Zagrobelny, M; Bak, S; Moller, BL. Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr. Opin. Plant. Biol 2005, 8, 280–291. [Google Scholar]
- Jovanović, S; Jovanović, A; Crawford, MR. m-ldh serves as a regulatory subunit of the cytosolic substrate-channelling complex in vivo. J. Mol. Biol 2007, 371, 349–361. [Google Scholar]
- Milani, M; Pesce, A; Bolognesi, M; Bocedi, A; Ascenzi, P. Substrate channeling: Molecular bases. Biochem. Mol. Biol. Ed 2003, 31, 228–233. [Google Scholar]
- Ishikawa, M; Tsuchiya, D; Oyama, T; Tsunaka, Y; Morikawa, K. Structural basis for channelling mechanism of a fatty acid bold beta-oxidation multienzyme complex. EMBO J 2004, 23, 2745–2754. [Google Scholar]
- Winkel, BS. Metabolic channeling in plants. Annu. Rev. Plant. Biol 2004, 55, 85–107. [Google Scholar]
- Barney, BM; Yurth, MG; Dos Santos, PC; Dean, DR; Seefeldt, LC. A substrate channel in the nitrogenase MoFe protein. J. Biol. Inorg. Chem 2009, 14, 1015–1022. [Google Scholar]
- Baker, P; Pan, D; Carere, J; Rossi, A; Wang, W; Seah, SY. Characterization of an aldolase-dehydrogenase complex that exhibits substrate channeling in the polychlorinated biphenyls degradation pathway. Biochemistry 2009, 48, 6551–6558. [Google Scholar]
- Grahama, JWA; Williamsa, TCR; Morgana, M; Fernieb, AR; Ratcliffea, RG; Sweetlovea, LJ. Glycolytic enzymes associate dynamically with mitochondria in response to respiratory demand and support substrate channeling. Plant Cell 2007, 19, 3723–3738. [Google Scholar]
- Commichau, FM; Rothe, FM; Herzberg, C; Wagner, E; Hellwig, D; Lehnik-Habrink, M; Hammer, E; Völker, U; Stülke, J. Novel activities of glycolytic enzymes in Bacillus subtilis: Interactions with essential proteins involved in mRNA processing. Mol. Cell. Proteomics 2009, 8, 1350–1360. [Google Scholar]
- Ngo, H; Kimmich, N; Harris, R; Niks, D; Blumenstein, L; Kulik, V; Barends, TR; Schlichting, I; Dunn, MF. Allosteric regulation of substrate channeling in tryptophan synthase: Modulation of the L-serine reaction in stage I of the beta-reaction by alpha-site ligands. Biochemistry 2007, 46, 7740–7753. [Google Scholar]
- Ovádi, J; Saks, V. On the origin of the ideas of intracellular compartmentation and organized metabolic systems. Mol. Cell. Biochem 2004, 256, 5–12. [Google Scholar]
- Ovádi, J; Orosz, F; Hollán, S. Functional aspects of cellular microcompartmentation in the development of neurodegeneration: Mutation induced aberrant protein-protein associations. Mol. Cell. Biochem 2004, 256–257, 83–93. [Google Scholar]
- Hudder, A; Nathanson, L; Deutscher, MP. Organization of mammalian cytoplasm. Mol. Cell. Biol 2003, 23, 9318–9326. [Google Scholar]
- Monge, C; Grichine, A; Rostovtseva, T; Sackett, P; Saks, VA. Compartmentation of ATP in cardiomyocytes and mitochondria. Kinetic studies and direct measurements. Biophys. J 2009, 96, 241a. [Google Scholar]
- Walter, H; Brooks, D; Srere, P. Microcompartmentation and Phase Separation in Cytoplasm; Academic Press: Orlando, FL, USA, 1999. [Google Scholar]
- Long, MS; Jones, CD; Helfrich, MR; Mangeney-Slavin, LK; Keating, CD. Dynamic microcompartmentation in synthetic cells. Proc. Natl. Acad. Sci.USA 2005, 102, 5920–5925. [Google Scholar]
- Lunn, EJ. Compartmentation in plant metabolism. J. Exp. Bot 2007, 58, 35–47. [Google Scholar]
- Saks, V; Khuchua, ZA; Vasilyeva, EV; OYu, B; Kuznetsov, AV. Metabolic compartmentation and substrate channelling in muscle cells. Role of coupled creatine kinases in in vivo regulation of cellular respiration-a synthesis. Mol. Cell. Biochem 1994, 133–134, 155–192. [Google Scholar]
- Saks, VA; Monge, C; Anmann, T; Dzeja, P. Saks, VA, Ed.; Integrated and organized cellular energeticsystems: Theories of cell energetics, compartmentation and metabolic channeling. In MolecularSystem Bioenergetics. Energy for Life; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Saks, V; Kaambre, T; Sikk, P; Eimre, M; Orlova, E; Paju, K; Piirsoo, A; Appaix, F; Kay, L; Regiz-Zagrosek, V; Fleck, E; Seppet, E. Intracellular energetic units in red muscle cells. Biochem. J 2001, 356, 643–665. [Google Scholar]
- Seppet, EK; Kaambre, T; Sikk, P; Tiivel, T; Vija, H; Tonkonogi, M; Sahlin, K; Kay, L; Appaix, F; Braun, U; Eimre, M; Saks, VA. Functional complexes of mitochondria with Ca, Mg, ATPases of myofibrils and sarcoplasmic reticulum in muscle cells. Biochim. Biophys. Acta 2001, 1504, 379–395. [Google Scholar]
- Kaasik, A; Veksler, V; Boehm, E; Novotova, M; Minajeva, A; Ventura-Clapier, R. Energetic crosstalk between organelles. Architectural integration of energy production and utilization. Circ. Res 2001, 89, 153–159. [Google Scholar]
- Saks, V; Beraud, N; Wallimann, T. Metabolic compartmentation–a system level property of muscle cells. Int. J. Mol. Sci 2008, 9, 751–767. [Google Scholar]
- Seppet, EK; Kaambre, T; Sikk, P; Tiivel, T; Vija, H; Tonkonogi, M; Sahlin, K; Kay, L; Appaix, F; Braun, U; Eimre, M; Saks, V. Functional Complexes of Mitochondria with Ca,Mg ATPases of Myofibrils and Sarcoplasmic Reticulum in Muscle Cells. Biochim. Biophys. Acta 2001, 1504, 379–395. [Google Scholar]
- Saks, V. Molecular System Bioenergetics; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Saks, V; Vendelin, M; Aliev, MK; Kekelidze, T; Engelbrecht, J. Dienel, G, Gibson, G, Eds.; Mechanisms and modeling of energy transfer between and among intracellular compartments. In Handbook of Neurochemistry and Molecular Neurobiology, 3rd ed; Springer Science and Business Media: Boston, NY, USA, 2007. [Google Scholar]
- Saks, V; Anmann, T; Guzun, R; Kaambre, T; Sikk, P; Schlattner, U; Wallimann, T; Aliev, M; Vendelin, M. Wyss, M, Salomons, G, Eds.; The creatine kinase phosphotransfer network: Thermodynamic and kinetic considerations, the impact of the mitochondrial outer membrane and modelling approaches. In Creatine and Creatine Kinase in Health and Disease; Springer: Dordrecht, The Netherlands, 2007. [Google Scholar]
- Monge, C; Beraud, N; Kuznetsov, AV; Rostovtseva, T; Sackett, D; Schlattner, U; Vendelin, M; Saks, VA. Regulation of respiration in brain mitochondria and synaptosomes: Restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol. Cell. Biochem 2008, 318, 147–165. [Google Scholar]
- Saks, V; Monge, C; Guzun, R. Philosophical basis and some historical aspects of systems biology: From hegel to noble - Applications for bioenergetic research. Int. J. Mol. Sci 2009, 10, 1161–1192. [Google Scholar]
- Yeates, TO; Tsai, Y; Tanaka, S; Sawaya, MR; Kerfeld, CA. Self-assembly in the carboxysome: A viral capsid-like protein shell in bacterial cells. Biochem. Soc. Trans 2007, 35, 508–511. [Google Scholar]
- Yeates, TO; Kerfeld, CA; Heinhorst, S; Cannon, GC; Shively, JM. Protein-based organelles in bacteria: Carboxysomes and related microcompartments. Nat. Rev. Microbiol 2008, 6, 681–691. [Google Scholar]
- Fan, C; Cheng, S; Liu, Y; Escobar, CM; Crowley, CS; Jefferson, RE; Yeates, TO; Bobik, TA. Short N-terminal sequences package proteins into bacterial microcompartments. Proc. Natl. Acad. Sci. USA 2010, 107, 7509–7514. [Google Scholar]
- Bobik, TA. Polyhedral organelles compartmenting bacterial metabolic processes. Appl. Microbiol. Biotechnol 2006, 70, 517–525. [Google Scholar]
- Yeates, TO; Kerfeld, CA; Heinhorst, S; Cannon, GC; Shively, JM. Protein-based organelles in bacteria: Carboxysomes and related microcompartments. Nat. Rev. Microbiol 2008, 6, 681–691. [Google Scholar]
- Sutter, M; Boehringer, D; Gutmann, S; Günther, S; Prangishvili, D; Loessner, MJ; Stetter, KO; Weber-Ban, E; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol 2008, 15, 939–947. [Google Scholar]
- Sampson, EM; Bobik, TA. Microcompartments for B12-dependent 1,2-propanediol degradation provide protection from DNA and cellular damage by a reactive metabolic intermediate. J. Bacteriol 2008, 190, 2966–2971. [Google Scholar]
- Goldbeter, A. Rythmes et chaos dans les systèmes biochímiques et cellulaires; Masson: Paris, France, 1990. [Google Scholar]
- Hess, B. Periodic patterns in biology. Naturwissenschaften 2000, 87, 199–211. [Google Scholar]
- Futcher, B. Metabolic cycle, cell cycle, and the finishing kick to start. Genome Biol 2006, 7, 107. [Google Scholar]
- Gillette, MU; Sejnowski, TJ. Biological clocks coordinately keep life on time. Science 2005, 309, 1196–1198. [Google Scholar]
- Hans, MA; Heinzle, E; Wittmann, C. Free intracellular amino acid pools during autonomous oscillations in Saccharomyces cerevisiae. Biotechnol. Bioeng 2003, 82, 143–151. [Google Scholar]
- Marquez, S; Crespo, P; Carlini, V; Garbarino-Pico, E; Baler, R; Caputto, BL; Guido, ME. The metabolism of phospholipids oscillates rhythmically in cultures of fibroblasts and is regulated by the clock protein PERIOD 1. FASEB J 2004, 18, 519–521. [Google Scholar]
- Hartig, K; Beck, E. Endogenous cytokinin oscillations control cell cycle progression of tobacco BY-2 Cells. Plant Biol 2005, 7, 33–40. [Google Scholar]
- Hungerbuehler, AK; Philippsen, P; Gladfelter, AS. Limited functional redundancy and oscillation of cyclins in multinucleated Ashbya gossypii fungal cells. Eukaryot. Cell 2007, 6, 473–486. [Google Scholar]
- Shaul, O; Mironov, V; Burssens, S; van Montagu, M; Inze, D. Two Arabidopsis cyclin promoters mediate distinctive transcriptional oscillation in synchronized tobacco BY-2 cells. Proc. Natl. Acad. Sci.USA 1996, 93, 4868–4872. [Google Scholar]
- Chabot, JR; Pedraza, JM; Luitel, P; van Oudenaarden, A. A Stochastic gene expression out-of-steady-state in the cyanobacterial circadian clock. Nature 2007, 450, 1249–1252. [Google Scholar]
- Tian, B; Nowak, DE; Brasier, AR. A TNF-induced gene expression program under oscillatory NF-κB control. BMC Genomics 2005, 6, 137. [Google Scholar]
- Tonozuka, H; Wang, J; Mitsui, K; Saito, T; Hamada, Y; Tsurugi, K. Analysis of the upstream regulatory region of the GTS1 gene required for its oscillatory expression. J. Biochem 2001, 130, 589–595. [Google Scholar]
- Klevecz, RR; Bolen, J; Forrest, G; Murray, DB. A genomewide oscillation in transcription gates DNA replication and cell cycle. Proc. Natl. Acad. Sci.USA 2004, 10, 1200–1205. [Google Scholar]
- Lange, G; Mandelkow, EM; Jagla, A; Mandelklow, E. Tubulin oligomers and microtubule oscillations Antagonistic role of microtubule stabilizers and destabilizers. FEBS Lett 2004, 178, 61–69. [Google Scholar]
- Placantonakis, DG; Welsh, JP. Two distinct oscillatory states determined by the NMDA receptor in rat inferior olive. J. Physiol 2001, 534, 123–140. [Google Scholar]
- De Forest, M; Wheeler, CJ. Coherent oscillations in membrane potential synchronize impulse bursts in central olfactory neurons of the crayfish. J. Neurophysiol 1999, 81, 1231–1241. [Google Scholar]
- Sánchez-Armáss, S; Sennoune, SR; Maiti, D; Ortega, F; Martínez-Zaguila, R. Spectral imaging microscopy demonstrates cytoplasmic pH oscillations in glial cells. Am. J. Physiol. Cell. Physiol 2006, 290, C524–C538. [Google Scholar]
- Holz, GG; Heart, E; Leech, CA. Synchronizing Ca2+ and cAMP oscillations in pancreatic beta cells: A role for glucose metabolism and GLP-1 receptors? Am. J. Physiol. Cell. Physiol 2008, 294, c4–c6. [Google Scholar]
- Ainscow, EK; Mirsham, S; Tang, T; Ashford, MLJ; Rutter, GA. Dynamic imaging of free cytosolic ATP concentration during fuel sensing by rat hypothalamic neurones: Evidence for ATPindependent control of ATP-sensitive K+ channels. J. Physiol 2002, 544, 429–445. [Google Scholar]
- Lloyd, D; Eshantha, L; Salgado, J; Turner, MP; Murray, DB. Respiratory oscillations in yeast: Clock-driven mitochondrial cycles of energization. FEBS Lett 2002, 519, 41–44. [Google Scholar]
- Rosenspire, AJ; Kindzelskii, AL; Petty, H. Pulsed DC electric fields couple to natural NAD(P)H oscillations in HT-1080 fibrosarcoma cells. J. Cell Sci 2001, 114, 1515–1520. [Google Scholar]
- Danù, S; Sùrensen, PG; Hynne, F. Sustained oscillations in living cells. Nature 1999, 402, 320–322. [Google Scholar]
- Ishii, K; Hirose, K; Iino, M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO 2006, 7, 390–396. [Google Scholar]
- Jules, M; Francois, J; Parrou, JL. Autonomous oscillations in Saccharomyces cerevisiae during batch cultures on trehalose. FEBS J 2005, 272, 1490–1500. [Google Scholar]
- Getty, L; Panteleon, AE; Mittelman, SD; Dea, MK; Bergman, RN. Rapid oscillations in omental lipolysis are independent of changing insulin levels in vivo. J. Clin. Invest 2000, 106, 421–430. [Google Scholar]
- Klevecz, RR; Murray, DB. Genome wide oscillations in expression – Wavelet analysis of time series data from yeast expression arrays uncovers the dynamic architecture of phenotype. Mol. Biol. Rep 2001, 28, 73–82. [Google Scholar]
- Brodsky, V; Boikov, PY; Nechaeva, NV; Yurovitsky, YG; Novikova, TE; Fateeva, VI; Shevchenko, NA. The rhythm of protein synthesis does not depend on oscillations of ATP level. J. Cell Sci 1992, 103, 363–370. [Google Scholar]
- Kindzelskii, AL; Zhou, MJ; Haugland, PR; Boxer, AL; Petty, RH. Oscillatory Pericellular Proteolysis and Oxidant Deposition During Neutrophil Locomotion. Biophys. J 1998, 74, 90–97. [Google Scholar]
- Fuentes, JM; Pascual, MR; Salido, G; Soler, JA. Oscillations in rat liver cytosolic enzyme activities of the urea cycle. Arch. Physiol. Biochem 1994, 102, 237–241. [Google Scholar]
- Wittmann, C; Hans, M; Van Winden, AW; Ras, C; Heijnen, JJ. Dynamics of intracellular metabolites of glycolysis and TCA cycle during cell-cycle-related oscillation in Saccharomyces cerevisiae. Biotechnol. Bioeng 2005, 89, 839–847. [Google Scholar]
- Aon, MA; Roussel, MR; Cortassa, S; O‘Rourke, B; Murray, DB; Beckmann, M; Lloyd, D. The scale-free dynamics of eukaryotic cells. Plos One 2008, 3. [Google Scholar]
- Garmendia-Torres, C; Goldbeter, A; Jacquet, M. Nucleocytoplasmic oscillations of the yeast transcription factor Msn2: Evidence for periodic PKA activation. Curr. Biol 2007, 17, 1044–1049. [Google Scholar]
- Barril, EF; Potter, AR. Systematic oscillations of amino acid transport in liver from rats adapted to controlled feeding schedules. J. Nutmtion 1968, 95, 228–237. [Google Scholar]
- M⊘ller, AC; Hauser, MJB; Olsen, LF. Oscillations in peroxidase-catalyzed reactions and their potential function in vivo. Biophys. Chem 1998, 72, 63–72. [Google Scholar]
- Smrcinova, M; S⊘rensen, PG; Krempasky, J; Ballo, P. Chaotic oscillations in a chloroplast system under constant illumination. Int. J. Bifurcat. Chaos 1998, 8, 2467–2470. [Google Scholar]
- Chiam, HK; Rajagopal, G. Oscillations in intracellular signaling cascades. Phys. Rev. E 2007, 75, 061901. [Google Scholar]
- Tu, BP; Kudlicki, A; Rowicka, M; McKnight, SL. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158. [Google Scholar]
- Goldbeter, A. Computational approaches to cellular rhythms. Nature 2002, 420, 238–245. [Google Scholar]
- Aon, MA; Cortassa, S; O‘Rourke, B. The fundamental organization of cardiac mitochondria as a network of coupled oscillators. Biophys. J 2006, 91, 4317–4327. [Google Scholar]
- Roussel, MR; Ivlev, AA; Igamberdiev, AU. Oscillations of the internal CO2 concentration in tobacco leaves transferred to low CO2. J. Plant Physiol 2006, 34, 1188–1196. [Google Scholar]
- Berridge, MJ; Galione, A. Cytosolic calcium oscillators. Faseb. J 1988, 2, 3074–3082. [Google Scholar]
- Chance, B; Pye, EK; Ghosh, AD; Hess, B. Biological and Biochemical Oscillations; Academic Press: New York, NY, USA, 1973. [Google Scholar]
- Brodsky, VY. Direct cell-cell communication: A new approach derived from recent data on the nature and self-organisation of ultradian (circahoralian) intracellular rhythms. Biol. Rev. Camb. Philos. Soc 2006, 81, 143–162. [Google Scholar]
- Dekhuijzen, A; Bagust, J. Analysis of neural bursting: Nonrhythmic and rhythmic activity in isolated spinal cord. J. Neurosci. Methods 1996, 67, 141–147. [Google Scholar]
- Olsen, LF; Degn, H. Chaos in biological systems. Q. Rev. Biophys 1985, 18, 165–225. [Google Scholar]
- Chandrashekaran, MK. Time in the Living World; Universities Press: Hyderabad, India, 2005. [Google Scholar]
- Lloyd, D; Murray, DB. Ultradian metronome: Timekeeper for orchestration of cellular coherence. Trends Biochem. Sci 2005, 30, 373–377. [Google Scholar]
- Lloyd, D; Murray, DB. The temporal architecture of eukaryotic growth. FEBS Lett 2006, 580, 2830–2835. [Google Scholar]
- Lloyd, D; Murray, DB. Redox rhythmicity: Clocks at the core of temporal coherence. Bioessays 2007, 29, 465–473. [Google Scholar]
- Hildebrandt, G. The Time Structure of Adaptive Processes; Georg Thieme Verlag: Stuttgart, Germany, 1982. [Google Scholar]
- Yates, FE. Boyd, CAR, Noble, D, Eds.; Self-organizing systems. In The Logic of Life; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Aon, MA; Cortassa, S. Dynamic biological organization. In Fundamentals as applied to cellular systems; Chapman & Hall: London, UK, 1997. [Google Scholar]
- Aon, MA; Cortassa, S; Lloyd, D. Chaotic dynamics and fractal space in biochemistry: Simplicity underlies complexity. Cell Biol. Int 2000, 24, 581–587. [Google Scholar]
- Wolf, J; Passarge, J; Somsen, OJG; Snoep, JL; Heinrich, R; Westerhof, HV. Transduction of intracellular and intercellular dynamics in yeast glycolytic oscillations. Biophys. J 2000, 78, 1145–1153. [Google Scholar]
- Tu, BP; Kudlicki, A; Rowicka, M; McKnight, SL. Logic of the yeast metabolic cycle: Temporal compartmentalization of cellular processes. Science 2005, 310, 1152–1158. [Google Scholar]
- Klevecz, RR; Bolen, J; Forrest, G; Murray, DB. A genomewide oscillation in transcription gates DNA replication and cell cycle. Proc. Natl. Acad. Sci. USA 2004, 101, 1200–1205. [Google Scholar]
- Murray, DB; Engelen, F; Lloyd, D; Kuriyama, H. Involvement of glutathione in the regulation of respiratory oscillation during a continuous culture of Saccharomyces cerevisiae. Microbiology 1999, 145, 2739–2745. [Google Scholar]
- Murray, DB; Klevecz, RR; Lloyd, D. Generation and maintenance of synchrony in Saccharomyces cerevisiae continuous culture. Exp. Cell. Res 2003, 287, 10–15. [Google Scholar]
- Klevecz, RR; Bolen, J; Forrest, G; Murray, DB. A genomewide oscillation in transcription gates DNA replication and cell cycle. Proc. Natl. Acad. Sci. USA 2004, 101, 1200–1205. [Google Scholar]
- Futcher, B. Metabolic cycle, cell cycle, and the finishing kick to Start. Genome Biol 2006, 7, 1–5. [Google Scholar]
- Oliva, A; Rosebrock, A; Ferrezuelo, F; Pyne, S; Chen, H; Skiena, S; Futcher, B; Leatherwood, J. The cell cycle-regulated genes of Schizosaccharomyces pombe. PLoS Biol 2005, 3, e225. [Google Scholar]
- Nelson, DE; Ihekwaba, AEC; Elliott, M; Johnson, JR; Gibney, CA; Foreman, BE; Nelson, G; See, V; Horton, CA; Spiller, DG; Edwards, SW; McDowell, HP; Unitt, JF; Sullivan, E; Grimley, R; Benson, N; Broomhead, D; Kell, DB; White, MRH. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar]
- Tian, B; Nowak, DE; Brasier, AR. A TNF-induced gene expression program under oscillatory NF- kappaB control. BMC Genomics 2005, 6, 137. [Google Scholar]
- Tonozuka, H; Wang, J; Mitsui, K; Saito, T; Hamada, Y; Tsurugi, K. Analysis of the upstream regulatory region of the GTS1 gene required for its oscillatory expression. J. Biochem 2001, 130, 589–595. [Google Scholar]
- Schibler, U; Sassone-Corsi, P. A web of circadian pacemakers. Cell 2002, 111, 919–922. [Google Scholar]
- Dunlap, JC; Loros, JJ; DeCoursey, P. Chronobiology: Biological Timekeeping; Sinauer Associates: Sunderland, MA, USA, 2004. [Google Scholar]
- Wijnen, H; Young, MW. Interplay of circadian clocks and metabolic rhythms. Annu. Rev. Genet 2006, 40, 409–448. [Google Scholar]
- Schibler, U; Naef, F. Cellular oscillators: Rhythmic gene expression and metabolism. Curr. Opin. Cell Biol 2005, 17, 223–229. [Google Scholar]
- Nakahata, Y; Grimaldi, B; Sahar, S; Hirayama, J; Sassone-Corsi, P. Signaling to the circadian clock: Plasticity by chromatin remodelling. Curr. Opin. Cell Biol 2007, 19, 230–237. [Google Scholar]
- Cardone, L; Hirayama, J; Giordano, F; Tamaru, T; Palvimo, JJ; Sassone-Corsi, P. Circadian clock control by SUMOylation of BMAL1. Science 2005, 309, 1390–1394. [Google Scholar]
- Hirayama, J; Sassone-Corsi, P. Structural and functional features of transcription factors controlling the circadian clock. Curr. Opin. Genet. Dev 2005, 15, 548–556. [Google Scholar]
- Dunlap, JC. Molecular bases for circadian clocks. Cell 1999, 96, 271–290. [Google Scholar]
- Corda, D; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J 2003, 22, 1953–1958. [Google Scholar]
- Freiman, RN; Tjian, R. Regulating the regulators: Lysine modifications make their mark. Cell 2003, 112, 11–17. [Google Scholar]
- Kojima, S; Hirose, M; Tokunaga, K; Sakaki, Y; Tei, H. Structural and functional analysis of 30 untranslated region of mouse Period1 mRNA. Biochem. Biophys. Res. Commun 2003, 301, 1–7. [Google Scholar]
- Baggs, JE; Green, CB. Nocturnin, a deadenylase in Xenopus laevis retina: A mechanism for posttranscriptional control of circadian-related mRNA. Curr. Biol 2003, 13, 189–198. [Google Scholar]
- Reppert, SM; Weaver, DR. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar]
- Martinek, S; Inonog, S; Manoukian, AS; Young, MW. A role for the segment polarity gene shaggy/GSK-3 in the Drosophila circadian clock. Cell 2001, 105, 769–779. [Google Scholar]
- Sathyanarayanan, S; Zheng, X; Xiao, R; Sehgal, A. Posttranslational regulation of Drosophila PERIOD protein by protein phosphatase 2A. Cell 2004, 116, 603–615. [Google Scholar]
- Lee, C; Etchegaray, JP; Cagampang, FR; Loudon, AS; Reppert, SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell 2001, 107, 855–867. [Google Scholar]
- Etchegaray, JP; Lee, C; Wade, PA; Reppert, SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 2003, 421, 177–182. [Google Scholar]
- Crosio, C; Cermakian, N; Allis, CD; Sassone-Corsi, P. Light induces chromatin modification in cells of the mammalian circadian clock. Nat. Neurosci 2000, 3, 1241–1247. [Google Scholar]
- Yagita, K; Tamanini, F; Yasuda, M; Hoeijmakers, JH; van der Horst, GT; Okamura, H. Nucleocytoplasmic shuttling and mCRYdependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J 2002, 21, 1301–1314. [Google Scholar]
- Gonze, D; Halloy, J; Goldbeter, A. Stochastic models for circadian oscillations: Emergence of a biological rhythm. Int. J. Quantum Chem 2004, 98, 228–238. [Google Scholar]
- Hunt, T; Sassone-Corsi, P. Circadian clocks and the cell cycle. Cell 2007, 129, 461–464. [Google Scholar]
- Granda, TG; Liu, X-H; Smaaland, R; Cermakian, N; Filipski, E; Sassone-Corsi, P; Lévi, F. Circadian regulation of cell cycle and apoptosis proteins in mouse bone marrow and tumor. FASEB J 2005, 19, 304–306. [Google Scholar]
- Petty, HR. Spatiotemporal chemical dynamics in living cells: From information trafficking to cell physiology. Biosystems 2006, 83, 217–224. [Google Scholar]
- Scemes, E; Giaume, C. Astrocyte calcium waves: What they are and what they do. Glia 2006, 54, 716–725. [Google Scholar]
- Galas, L; Garnier, M; Lamacz, M. Calcium waves in frog melanotrophs are generated by intracellular inactivation of TTX-sensitive membrane Na_ channel. Mol. Cell. Endocrinol 2000, 170, 197–209. [Google Scholar]
- Guthrie, PB; Knappenberger, J; Segal, M; Bennett, MVL; Charles, AC; Kater, SB. ATP released from astrocytes mediates glial calcium waves. J. Neurosci 1999, 19, 520–528. [Google Scholar]
- Bernardinelli, Y; Magistretti, PJ; Chatton, J-Y. Astrocytes generate Na+-mediated metabolic waves. Proc. Natl. Acad. Sci. USA 2004, 101, 14937–14942. [Google Scholar]
- Romashko, DN; Marban, E; O‘Rourke, B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1618–1623. [Google Scholar]
- Ueda, T; Nakagaki, T; Yamada, T. Dynamic organization of ATP and birefringent fibrils during free locomotion and galvanotaxis in the plasmodium of Physarum polycephalum. J. Cell. Biol 1990, 110, 1097–1102. [Google Scholar]
- Newman, EA. Propagation of intercellular calcium waves in retinal astrocytes and müller cells. J. Neurosci 2001, 21, 2215–2223. [Google Scholar]
- Petty, HR; Worth, RG; Kindzelskii, AL. Imaging sustained dissipative patterns in the metabolism of individual living cells. Phys. Rev. Lett 2000, 84, 2754–2757. [Google Scholar]
- Petty, HR; Kindzelskii, AL. Dissipative metabolic patterns respond during neutrophil transmembrane signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3145–3149. [Google Scholar]
- Slaby, O; Lebiedz, D. Oscillatory NAD(P)H waves and calcium oscillations in neutrophils? A modeling study of feasibility. Biophys. J 2009, 96, 417–428. [Google Scholar]
- Vicker, MG. Eukaryotic cell locomotion depends on the propagation of self-organized reaction-diffusion waves and oscillations of actin filament assembly. Exp. Cell. Res 2002, 275, 54–66. [Google Scholar]
- Asano, Y; Nagasaki, A; Uyeda, TQP. Correlated waves of actin filaments and PIP3 in Dictyostelium cells. Cell Mot. Cytosk 2008, 65, 923–934. [Google Scholar]
- De La Fuente, IM; Benítez, N; Santamaría, A; Veguillas, J; Aguirregabiria, JM. Persistence in metabolic nets. Bull. Mathemat. Biol 1999, 61, 573–595. [Google Scholar]
- Almaas, E; Kovacs, B; Vicsek, T; Oltvai, ZN; Barabási, AL. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 2004, 427, 839–843. [Google Scholar]
- Almaas, E; Oltvai, ZN; Barabasi, AL. The activity reaction core and plasticity of metabolic networks. PLoS Comput. Biol 2005, 1. [Google Scholar]
- Almaas, E. Biological impacts and context of network theory. J. Exp. Biol 2007, 210, 1548–1558. [Google Scholar]
- De La Fuente, IM; Martínez, L; Pérez-Samartín, AL; Ormaetxea, L; Amezaga, C; Vera-López, A. Global self-organization of the cellular metabolic structure. Plos One 2008, 3, e3100:1–e3100:19. [Google Scholar]
- De La Fuente, IM; Vadillo, F; Pérez-Pinilla, M-B; Vera-López, A; Veguillas, J. The number of catalytic elements is crucial for the emergence of metabolic cores. Plos One 2009, 4. [Google Scholar]
- De La Fuente, IM; Vadillo, F; Pérez-Samartín, AL; Pérez-Pinilla, M-B; Bidaurrazaga, J; Vera-López, A. Global self-regulations of the cellular metabolic structure. Plos One 2010, 5, e9484:1–e9484:15. [Google Scholar]
- Prigogine, I; Mayné, F; George, C; De Haan, M. Microscopic theory of irreversible processes. Proc. Nat. Acad. Sci. USA 1977, 74, 4152–4156. [Google Scholar]
- Glansdorf, P; Prigogine, I. Thermodynamic Theory of Structure, Stability, and Fluctuations; Wiley Interscience: New York, NY, USA, 1971. [Google Scholar]
- Ebeling, W; Engel-Herbert, H; Herzel, H. Ebeling, W, Ulbricht, H, Eds.; Thermodynamic aspects of selforganization. In Selforganization by Non-Linear Irreversible Processes; Springer-Verlag: Berlin, Germany, 1986. [Google Scholar]
- Klimontovich, YL. Entropy and information of open systems. Phys.-Uspekhi 1999, 42, 375–384. [Google Scholar]
- Karsenti, E. Self-organization in cell biology: A brief history. Nat. Rev. Mol. Cell Biol 2008, 9, 255–262. [Google Scholar]
- van de Vijver, G. Self-organization and Emergence in Life Sciences; Springer: Dordrecht, Holland, 2006. [Google Scholar]
- Wicken, JS. Information transformation in molecular evolution. J. Theor. Biol 1978, 72, 191–204. [Google Scholar]
- Wicken, J. The generation of complexity in evolution: A thermodynamic and information-theoretical discussion. J. Theor. Biol 1979, 77, 349–365. [Google Scholar]
- Pulselli, RM; Simoncini, E; Tiezzi, E. Self-organization in dissipative structures: A thermodynamic theory for the emergence of prebiotic cells and their epigenetic evolution. BioSystems 2009, 96, 237–241. [Google Scholar]
- Sidelnikov, DI; Gritsenko, OV; Simonova, AP; Rambidi, NG. Nonlinear dynamics of the distributed biochemical systems functioning in the dissipative structure formation mode. Biol. Cybern 1992, 68, 53–62. [Google Scholar]
- Micheaua, JC; de Mina, M; Gimeneza, M. Dissipative structures and amplification of enantiomeric excess (an experimental point of view). Biosystems 1987, 20, 85–93. [Google Scholar]
- Ebeling, W. On the entropy of dissipative and turbulent structures. Phys. Scr 1989, T25, 238–242. [Google Scholar]
- Li, M; Vitanyi, P. An Introduction to Kolmogorov Complexity and Its Applications; Springer: New York, NY, USA, 1997. [Google Scholar]
- Surrey, T; Nedelec, F; Leibler, S; Karsenti, E. Physical properties determining self-organization of motors and microtubules. Science 2001, 292, 1167–1171. [Google Scholar]
- Pohl, K; Bartelt, MC; de la Figuera, J; Bartelt, NC; Hrbek, J; Hwang, RQ. Identifying the forces responsible for self-organization of nanostructures at crystal surfaces. Nature 1998, 1999, 238–241. [Google Scholar]
- Papaseit, C; Pochon, N; Tabony, J. Microtubule self-organization is gravity-dependent. Proc. Natl. Acad. Sci. USA 2000, 97, 8364–8368. [Google Scholar]
- Kurakin, A. Self-organization versus watchmaker: Molecular motors and protein translocation. Biosystems 2006, 84, 15–23. [Google Scholar]
- Whitesides, GM; Boncheva, M. Supramolecular chemistry and self-assembly special feature: Beyond molecules: Self-assembly of mesoscopic and macroscopic components. Proc. Nat. Acad. Sci. USA 2002, 99, 4769–4774. [Google Scholar]
- Orgel, LE. Self-organizing biochemical cycles. Proc. Natl. Acad. Sci. USA 2000, 97, 12503–12507. [Google Scholar]
- Fletcher, G; Mason, S; Terrett, J; Soloviev, M. Self-assembly of proteins and their nucleic acids. J. Nanobiotechnol 2003, 1, 1. [Google Scholar]
- Yockey, HP. Information Theory, Evolution, and the Origin o Life; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Pattee, HH. Andersen, PB, Emmeche, C, Finnemann, NO, Christiansen, PV, Eds.; Causation, control, and the evolution of complexity. In Downward Causation: Minds, Bodies, and Matter; Aarhus University Press: Aarhus, DK, USA, 2000. [Google Scholar]
- Kauffman, S. The Origins of Order: Self-Organization and Selection in Evolution; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Kauffman, S. At Home in the Universe: The Search for the Laws of Self-Organization and Complexity; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Duysens, LNM; Amesz, J. Fluorescence espectrophotometry of reduced phosphopyridine nucleotide in intact cells in the near-ultraviolet and visible region. Biochem. Biophys. Acta 1957, 24, 19. [Google Scholar]
- Chance, B; Hess, B; Betz, A. DPNH oscillations in a cell-free extract of S. carlsbergensis. Biochem. Biophys. Res. Commun 1964, 16, 182–187. [Google Scholar]
- Higgins, J. A chemical mechanism for oscillation of glycolytic intermediates in yeast cells. Proc. Natl. Acad. Sci. USA 1964, 51, 988–994. [Google Scholar]
- Selkov, EE. Self-oscillations in glycolysis. Eur. J. Biochem 1968, 4, 79–86. [Google Scholar]
- Goldbeter, A; Lefeber, R. Disipative estructures for an allosteric model. Biophys. J 1972, 12, 1302–1315. [Google Scholar]
- Goldbeter, A. Patterns of spatiotemporal organization in an allosteric enzyme model. Proc. Natl. Acad. Sci. USA 1973, 70, 3255–3259. [Google Scholar]
- Goldbeter, A. Modulation of the adenylate energy charge by sustained metabolic oscillations. Febs. Lett 1974, 43, 327–330. [Google Scholar]
- Boiteux, A; Goldbeter, A; Hess, B. Control of oscillating glycolysis of yeast by stochastic, periodic, and steady source of substrate: A model and experimental study. Proc. Natl. Acad. Sci. USA 1975, 72, 3829–3833. [Google Scholar]
- Decroly, O; Goldbeter, A. Birhytmicity, chaos and other patterns of temporal self-organization in a multiply regulated biochemical system. Proc. Natl. Acad. Sci. USA 1982, 79, 6917–6921. [Google Scholar]
- Monod, J; Wyman, J; Changeux, JP. On the nature of allosteric transitions: A plausible model. J. Mol. Biol 1965, 12, 88–118. [Google Scholar]
- Mogilevskaya, E; Demin, O; Goryanin, I. Kinetic Model of Mitochondrial krebs cycle: Unraveling the mechanism of salicylate hepatotoxic effects. J. Biol. Phys 2006, 32, 245–271. [Google Scholar]
- Yang, CR; Shapiro, BE; Hung, SP; Mjolsness, ED; Hatfield, GW. A mathematical model for the branched chain amino acid biosynthetic pathways of Escherichia coli K12. J. Biol. Chem 2005, 280, 11224–11232. [Google Scholar]
- Korzeniewski, B; Zoladz, JA. A model of oxidative phosphorylation in mammalian skeletal muscle. Biophys. Chem 2001, 92, 17–34. [Google Scholar]
- Korzeniewski, B; Zoladz, JA. A model of oxidative phosphorylation in mammalian skeletal muscle. Biophys. Chem 2001, 92, 17–34. [Google Scholar]
- Bier, M; Teusink, B; Kholodenko, BN; Westerhoff, HV. Control analysis of glycolytic oscillations. Biophys. Chem 1996, 62, 15–24. [Google Scholar]
- Kass, L; Bray, WO. Kinetic model for phototransduction and G-protein enzyme cascade: Understanding quantal bumps during inhibition of CaM-KII or PP2B. J. Photochem. Photobiol. B: Biol 1996, 35, 105–113. [Google Scholar]
- Smidtas, S; Schächter, V; Képès, F. The adaptive filter of the yeast galactose pathway. J. Theor. Biol 2006, 242, 372–381. [Google Scholar]
- Petrov, V; Peifer, M; Timmer, J. Bistability and self-oscillations in cell cycle control. Int. J. Bifurc. Chaos 2006, 16, 1057–1066. [Google Scholar]
- Groenenboom, MAC; Marée, AFM; Hogeweg, P. The RNA silencing pathway: The bits and pieces that matter. PLoS Comput. Biol 2005, 1, e21. [Google Scholar]
- Reidl, J; Borowski, P; Sensse, A; Starke, J; Zapotocky, M; Eiswirthz, M. Model of calcium oscillations due to negative feedback in olfactory cilia. Biophys. J 2006, 90, 1147–1155. [Google Scholar]
- Wawra, C; Kuhl, M; Kestler, HA. Extended analyses of the Wnt/b-catenin pathway: Robustness and oscillatory behaviour. FEBS Lett 2007, 581, 4043–4048. [Google Scholar]
- Bergmann, SR; Weinheimer, CJ; Markham, J; Herrero, P. Quantitation of myocardial fatty acid metabolism using PET. J. Nucl. Med 1996, 37, 1723–1730. [Google Scholar]
- Sokhansanj, BA; Rodrigue, GR; Fitch, JP; Wilson, DM. A quantitative model of human DNA base excision repair. I. Mechanistic insights. Nucleic. Acids. Res 2002, 30, 1817–1825. [Google Scholar]
- Smieja, J; Jamaluddin, M; Brasier, AR; Kimmel, M. Model-based analysis of interferon-β induced signaling pathway. Bioinformatics 2008, 24, 2363–2369. [Google Scholar]
- Cheong, R; Levchenko, A. Wires in the soup: Quantitative models of cell signaling. Trends Cell Biol 2008, 18, 112–118. [Google Scholar]
- Alvarez-Vasquez, F; Sims, KS; Cowart, LA; Okamoto, Y; Voit, EO; Hannun, YA. Simulation and validation of modelled sphingolipid metabolism in Saccharomyces cerevisiae. Nature 2005, 433, 425–430. [Google Scholar]
- Yang, J; Clark, JW; Bryan, RM; Robertson, CS. Mathematical modeling of the nitric oxide/cGMP pathway in the vascular smooth muscle cell. Am. J. Physiol. Heart Circ. Physiol 2005, 289, H886–H897. [Google Scholar]
- Tyson, JJ. Modeling the cell division cycle: CDC2 and cyclin interactions. Proc. Natl. Acad. Sci. USA 1991, 88, 7328–7332. [Google Scholar]
- Novak, B; Tyson, JJ. Numerical analysis of a comprehensive model of M-phase control in Xenopus oocyte extracts and intact embryos. J. Cell Sci 1993, 106, 1153–1168. [Google Scholar]
- Tyson, J; Novaki, B; Odell, MG; Chen, K; Thron, CD. Chemical kinetic theory: Understanding cell-cycle regulation. TIBS 1996, 21, 89–96. [Google Scholar]
- Sha, W; Moore, J; Chen, K; Lassaletta, AD; Yi, CS; Tyson, JJ; Sible, JC. From the Cover: Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl. Acad. Sci. USA 2003, 100, 975–980. [Google Scholar]
- Pomerening, JR; Sontag, ED; Ferrell, JE, Jr. Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell. Biol 2003, 5, 346–351. [Google Scholar]
- Pomerening, JR; Kim, SY; Ferrell, JE, Jr. Systems-level dissection of the cell-cycle oscillator: Bypassing positive feedback produces damped oscillations. Cell 2005, 122, 565–578. [Google Scholar]
- Kapuy, O; He, E; Lopez-Avilies, S; Uhlmann, F; Tyson, JJ; Novak, B. System-level feedbacks control cell cycle progression. FEBS Lett 2009, 583, 3992–3998. [Google Scholar]
- Pomerening, JR; Sontag, ED; Ferrell, JE, Jr. Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol 2003, 5, 346–351. [Google Scholar]
- Kar, S; Baumann, WT; Paul, MR; Tyson, JJ. Exploring the roles of noise in the eukaryotic cell cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 6471–6476. [Google Scholar]
- Csikasz-Nagy, A; Kapuy, O; Toth, A; Pal, C; Jensen, LJ; Uhlmann, F; Tyson, JJ; Novak, B. Cell cycle regulation by feed-forward loops coupling transcription and phosphorylation. Mol. Syst. Biol 2009, 5, 236. [Google Scholar]
- Heinrich, R; Schuster, S. The Regulation of Cellular Systems; Chapman & Hall: New York, NY, USA, 1996. [Google Scholar]
- Reisig, W. Petri Nets: An Introduction; Springer: Berlin, Germany, 1985. [Google Scholar]
- Reddy, VN; Liebman, MN; Mavrovouniotis, ML. Qualitative analysis of biochemical reaction systems. Comp. Biol. Med 1996, 26, 924–935. [Google Scholar]
- Schuster, S; Pfeiffer, T; Moldenhauer, F; Koch, I; Dandekar, T. Exploring the pathway structure of metabolism: Decomposition into subnetworks and application to Mycoplasma pneumoniae. Bioinformatics 2002, 18, 351–361. [Google Scholar]
- Zevedei-Oancea, I; Schuster, S. Topological analysis of metabolic networks based on Petri net theory. Silico Biol 2003, 3, 323–345. [Google Scholar]
- Kwang-Il, G; Eulsik, O; Hawoong, J; Byungnam, K; Kim, D. Classification of scale-free networks. Proc. Natl. Acad. Sci. USA 2002, 99, 12583–12588. [Google Scholar]
- Albert, R. Scale-free networks in cell biology. J. Cell. Sci 2005, 118, 4947–4957. [Google Scholar]
- Newman, MEJ. Models of the small world. J. Stat. Phys 2000, 101, 819–841. [Google Scholar]
- Barabási, AL; Oltvai, ZN. Network biology: Understanding the Cells's functional organization. Nat. Rev. Genet 2004, 5, 101–113. [Google Scholar]
- Bernhardsson, S; Minnhagen, P. Models and average properties of scale-free directed networks. Phys. Rev. E 2006, 74, 026104. [Google Scholar]
- Raman, K; Rajagopalan, P; Chandra, N. Flux balance analysis of mycolic acid pathway: Targets for anti-tubercular drugs. PLoS Comput. Biol 2005, 1, e46. [Google Scholar]
- Ma, HW; Zhao, XM; Yuan, YJ; Zeng, AP. Decomposition of metabolic network based on the global connectivity structure of reaction graph. Bioinformatics 2004, 12, 1870–1876. [Google Scholar]
- Kim, DH; Rodgers, GJ; Kahng, B; Kim, D. Modelling hierarchical and modular complex networks: Division and independence. Phys. A 2005, 351, 671–679. [Google Scholar]
- Chen, F; Chen, Z; Liu, Z; Xiang, L; Yuan, Z. Finding and evaluating the hierarchical structure in complex networks. J. Phys. A: Math. Theor 2007, 40, 5013–5023. [Google Scholar]
- Feist, AM; Palsson, BO. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat. Biotechnol 2008, 26, 659–667. [Google Scholar]
- Feist, AM; Henry, CS; Reed, JL; Krummenacker, M; Joyce, AR; Karp, PD; Broadbelt, LJ; Hatzimanikatis, V; Palsson, B. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol 2007, 3, 121. [Google Scholar]
- Duarte, NC; Herrgard, MJ; Palsson, BO. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res 2004, 14, 1298–1309. [Google Scholar]
- Forster, J; Famili, I; Fu, P; Palsson, BO; Nielsen, J. Genome-scale reconstruction of the Saccharomyces cerevisiae metabolic network. Genome Res 2003, 13, 244–253. [Google Scholar]
- Herrgard, MJ; Swainston, N; Dobson, P; Dunn, WB; Arga, KY; Arvas, M; Büthgen, N; Borger, S; Costenoble, R; Heinemann, M; Hucka, M; Le Novère, N; Li, P; Liebermeister, W; Mo, ML; Oliveira, AP; Petranovic, D; Pettifer, S; Simeonidis, E; Smallbone, K; Spasié, I; Weichart, D; Brent, R; Broomhead, DS; Westerhoff, HV; Kürdar, B; Penttilä, M; Klipp, E; Palsson, B; Sauer, U; Oliver, SG; Mendes, P; Nielsen, J; Kell, DB. A consensus yeast metabolic network reconstruction obtained from a community approach to systems biology. Nat. Biotechnol 2008, 26, 1155–1160. [Google Scholar]
- Duarte, NC; Becker, SA; Jamshidi, N; Thiele, I; Mo, ML; Vo, TD; Srivas, R; Palsson, B. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar]
- Ma, H; Sorokin, A; Mazein, A; Selkov, A; Selkov, E; Demin, O; Goryanin, I. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol. Syst. Biol 2007, 3, 135. [Google Scholar]
- Burgard, AP; Nikolaev, EV; Schilling, CH; Maranas, CD. Flux coupling analysis of genome-scale metabolic reconstructions. Genome Res 2004, 14, 301–312. [Google Scholar]
- Reed, JL; Palsson, BO. Genome-scale in silico models of E. coli have multiple equivalent phenotypic states: Assessment of correlated reaction subsets that comprise network states. Genome Res 2004, 14, 1797–1805. [Google Scholar]
- Lee, JM; Gianchandani, WP; Papin, JA. Flux balance analysis in the era of metabolomics. Brief. Bioinform 2006, 7, 140–150. [Google Scholar]
- Feist, AM; Herrgard, MJ; Thiele, I; Reed, JL; Palsson, BO. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol 2009, 7, 129–143. [Google Scholar]
- Gianchandani, EP; Brautigan, DL; Papin, JA. Systems analyses characterize integrated functions of biochemical networks. Trends Biochem. Sci 2006, 31, 284–291. [Google Scholar]
- Schilling, CH; Covert, MW; Famili, I; Church, GM; Edwards, JS; Palsson, B. Genome-scale metabolic model of Helicobacter pylori 26695. J. Bacteriol 2002, 184, 4582–4593. [Google Scholar]
- Reed, JL; Vo, TD; Schilling, CH; Palsson, B. An expanded genomescale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 2003, 4, R54.1–R54.12. [Google Scholar]
- Duarte, NC; Herrgard, MJ; Palsson, B. Reconstruction and validation of Saccharomyces cerevisiae iND750, a fully compartmentalized genome-scale metabolic model. Genome Res 2004, 14, 1298–1309. [Google Scholar]
- Feist, AM; Herrgard, MJ; Thiele, I; Reed, JL; Palsson, B. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol 2009, 7, 129–143. [Google Scholar]
- Gianchandani, EP; Brautigan, DL; Papin, JA. Systems analyses characterize integrated functions of biochemical networks. Trends Biochem. Sci 2006, 31, 284–291. [Google Scholar]
- Lee, JM; Gianchandani, EP; Papin, JA. Flux balance analysis in the era of metabolomics. Brief. Bioinform 2006, 7, 140–150. [Google Scholar]
- Price, ND; Papin, JA; Schilling, CH; Palsson, B. Genome-scale microbial in silico models: The constraints-based approach. Trends Biotechnol 2003, 21, 162–169. [Google Scholar]
- Gianchandani, EP; Oberhardt, MA; Burgard, AP; Maranas, CD; Papin, JA. Predicting biological system objectives de novo from internal state measurements. BMC Bioinform 2008, 9, 43. [Google Scholar]
- Varma, A; Palsson, B. Metabolic flux balancing—basic concepts, scientific and practical use. Nat. Biotechnol 1994, 12, 994–998. [Google Scholar]
- Pramanik, J; Keasling, JD. Stoichiometric model of Escherichia coli metabolism: Incorporation of growth-rate dependent biomass composition and mechanistic energy requirements. Biotech. Bioeng 1997, 56, 398–421. [Google Scholar]
- Edwards, JS; Ibarra, RU; Palsson, B. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotechnol 2001, 19, 125–130. [Google Scholar]
- Blank, LM; Ebert, BE; Buhler, B; Schmid, A. Metabolic capacity estimation of Escherichia coli as a platform for redox biocatalysis: Constraint-based modeling and experimental verification. Biotechnol. Bioeng 2008, 100, 1050–1065. [Google Scholar]
- Portnoy, VA; Herrgard, MJ; Palsson, B. Aerobic fermentation of D-glucose by an evolved cytochrome oxidase-deficient Escherichia coli strain. Appl. Environ. Microbiol 2008, 74, 7561–7569. [Google Scholar]
- Lee, KH; Park, JH; Kim, TY; Kim, HU; Lee, SY. Systems metabolic engineering of Escherichia coli for L-threonine production. Mol. Syst. Biol 2007, 3, 149. [Google Scholar]
- Izallalen, M; Mahadevan, R; Burgard, A; Postier, B; Didonato, R, Jr; Sun, J; Schilling, CH; Lovley, DR. Geobacter sulfurreducens strain engineered for increased rates of respiration. Metab. Eng 2008, 10, 267–275. [Google Scholar]
- Cakir, T; Efe, C; Dikicioglu, D; Hortacsu, A; Kirdar, B; Oliver, SG. Flux balance analysis of a genome-scale yeast model constrained by exometabolomic data allows metabolic system identification of genetically different strains. Biotechnol. Prog 2007, 23, 320–326. [Google Scholar]
- Vo, TD; Greenberg, HJ; Palsson, B. Reconstruction and functional characterization of the human mitochondrial metabolic network based on proteomic and biochemical data. J. Biol. Chem 2004, 279, 39532–39540. [Google Scholar]
- Covert, MW; Schilling, CH; Palsson, B. Regulation of gene expression in flux balance models of metabolism. J. Theor. Biol 2001, 213, 73–88. [Google Scholar]
- Covert, MW; Palsson, B. Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J. Biol. Chem 2002, 277, 28058–28064. [Google Scholar]
- Covert, MW; Palsson, B. Constraints-based models: Regulation of gene expression reduces the steady-state solution space. J. Theor. Biol 2003, 221, 309–325. [Google Scholar]
- Beard, DA; Liang, SD; Qian, H. Energy balance for analysis of complex metabolic networks. Biophys. J 2002, 83, 79–86. [Google Scholar]
- Gianchandani, EP; Papin, JA; Price, ND; Joyce, AR; Palsson, B. Matrix formalism to describe functional states of transcriptional regulatory systems. PLoS Comput. Biol 2006, 2, e101. [Google Scholar]
- Mahadevan, R; Edwards, JS; Doyle, FJ, III. Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys. J 2002, 83, 1331–1340. [Google Scholar]
- Lee, JM; Gianchandani, EP; Eddy, JA; Papin, JA. Dynamic analysis of integrated signaling, metabolic, and regulatory networks. PLoS Comput. Biol 2008, 4, e1000086. [Google Scholar]
- Hess, B; Boiteux, A; Kruger, J. Cooperation of glycolytic enzymes. Adv. Enzyme Regul 1969, 7, 149–167. [Google Scholar]
- Hess, B; Boiteux, A. Oscillatory phenomena in biochemistry. Annu. Rev. B: Chem 1971, 40, 237–258. [Google Scholar]
- De la Fuente, MI; Martínez, L; Veguillas, J. Dynamic behavior in glycolytic oscillations with phase shifts. Biosystems 1995, 35, 1–13. [Google Scholar]
- De la Fuente, MI; Martínez, L; Veguillas, J. Intermittency route to chaos in a biochemical system. Biosystems 1996, 39, 87–92. [Google Scholar]
- De la Fuente, MI; Martínez, L; Veguillas, J; Aguirregabiria, JM. Quasiperiodicity route to chaos in a biochemical system. Biophys. J 1996, 71, 2375–2379. [Google Scholar]
- De la Fuente, MI; Martínez, L; Veguillas, J; Aguirregabiria, JM. Coexistence of multiple periodic and chaotic regimes in biochemical oscillations. Acta Biotheor 1996, 46, 37–51. [Google Scholar]
- De la Fuente, MI. Diversity of temporal self-organized behaviors in a biochemical system. BioSystems 1999, 50, 83–97. [Google Scholar]
- Viola, ER; Raushel, MF; Rendina, RA; Cleland, WW. Substrate synergism and the kinetic mechanism of yeast hexokinase. Biochemistry 1982, 21, 1295–1302. [Google Scholar]
- Goldbeter, A. Modulation of the adenylate energy charge by sustained metabolic oscillations. Febs Lett 1974, 43, 327–330. [Google Scholar]
- Boiteux, A; Goldbeter, A; Hess, B. Control of oscillating glycolysis of yeast by stochastic, periodic, and steady source of substrate: A model and experimental study. Proc. Natl. Acad. Sci. USA 1975, 72, 3829–3833. [Google Scholar]
- Markus, M; Plesser, T; Boiteux, A; Hess, B; Malcovati, M. Rate law of pyruvate kinase type I from Escherichia coli. Biochem. J 1980, 189, 421–433. [Google Scholar]
- Goldbeter, A; Lefever, R. Dissipative structures for an allosteric model. Biophys. J 1972, 12, 1302–1314. [Google Scholar]
- Laurent, M; Dessen, P; Seydoux, JF. Allosteric regulation of yeast phosphofructokinase. J. Biol. Chem 1979, 254, 7515–7520. [Google Scholar]
- Hess, B; Kuschmitz, D; Markus, M. Richard, J, Cornish-Bowden, A, Eds.; Dynamics of Biochemical Systems; Plenum: New York, NY, USA, 1984. [Google Scholar]
- Markus, M; Muler, SC; Hess, B. Observation of entertainment quasiperiodicity and chaos in glycolyzing yeast extracts under periodic glucose input. Ber. Bunsengues. Phys. Chem 1985, 89, 651. [Google Scholar]
- Hess, B; Markus, M; Muler, SC; Plesser, T. Gray, P, Nicolis, G, Baras, F, Borckmans, P, Scott, SK, Eds.; From homogeneity towards the anatomy of a chemical spiral. In Spatial Inhomogeneities and Transient Behavior in Chemical Kinetics; Manchester University Press: Manchester, UK, 1990. [Google Scholar]
- Bartrons, R; Schaftingen, E; Vissers, S; Hers, H. The stimulation of yeast phosphofructokinase by fructose 2,6-bisphosfate. FEBS Lett 1982, 143, 137–140. [Google Scholar]
- Ruelle, D; Takens, F. On the nature of turbulence. Commun. Math. Phys 1971, 20, 167–172. [Google Scholar]
- Newhouse, S; Ruelle, D; Takens, F. Occurrence of strange axiom-A attractors near quasiperiodic flow on Tm, m > 3. Commun. Math. Phys 1978, 64, 35–44. [Google Scholar]
- Rustici, M; Branca, M; Brunetti, A; Caravati, C; Marchettini, N. Inverse Ruelle-Takens- Newhouse scenario in a closed unstirred cerium-catalysed Belousov-Zhabotinsky system. Chem. Phys. Lett 1998, 293, 145–151. [Google Scholar]
- Hasty, J; McMillen, D; Isaacs, F; Collins, JJ. Computational studies of gene regulatory networks: In numero molecular biology. Nat. Rev. Genet 2001, 2, 268–279. [Google Scholar]
- Kærn, M; Elston, TC; Blake, WJ; Collins, JJ. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet 2005, 6, 451–464. [Google Scholar]
- Smolen, P; Baxter, DA; Byrne, JH. Mathematical modeling of gene networks. Neuron 2000, 26, 567–580. [Google Scholar]
- Ferrell, JE, Jr. Self-perpetuating states in signal transduction: Positive feedback, double-negative feedback and bistability. Curr. Opin. Cell Biol 2002, 14, 140–148. [Google Scholar]
- Slepchenko, BM; Terasaki, M. Bio-switches: What makes them robust? Curr. Opin. Genet. Dev 2004, 14, 428–434. [Google Scholar]
- Ptashne, MA. Genetic Switch: Phage Lambda and Higher Organisms; Cell: Cambridge, MA, USA, 1992. [Google Scholar]
- McAdams, HH; Arkin, A. Stochastic mechanisms in gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 814–819. [Google Scholar]
- Arkin, A; Ross, J; McAdams, HH. Stochastic Kinetic Analysis of Developmental Pathway Bifurcation in Phage {lambda}-Infected Escherichia coli Cells. Genetics 1998, 149, 1633–1648. [Google Scholar]
- Issacs, FJ; Hasty, J; Cantor, CR; Collins, JJ. Prediction and measurement of an autoregulatory genetic module. Proc. Natl. Acad. Sci. USA 2003, 100, 7714–7719. [Google Scholar]
- Gardner, TS; Cantor, CR; Collins, JJ. Construction of a genetic toggle switch in Escherichia coli. Nature 2000, 403, 339–342. [Google Scholar]
- Kobayashi, H; Kærn, M; Araki, M; Chung, K; Gardner, TS; Cantor, CR; Collins, JJ. Programmable cells: Interfacing natural and engineered gene networks. Proc. Natl. Acad. Sci. USA 2004, 101, 8414–8419. [Google Scholar]
- Kramer, BP; Viretta, AU; El Baba, MD; Aubel, D; Weber, W; Fussenegger, M. An engineered epigenetic transgene switch in mammalian cells. Nat. Biotechnol 2004, 22, 867–870. [Google Scholar]
- Novick, A; Wiener, M. Enzyme induction as an all-or-none phenomenon. Proc. Natl. Acad. Sci. USA 1957, 43, 553–566. [Google Scholar]
- Vilar, JMG; Guet, CC; Leibler, S. Modeling network dynamics: The lac operon, a case study. J. Cell Biol 2003, 161, 471–476. [Google Scholar]
- Ozbudak, EM; Thattai, M; Lim, HN; Shraiman, BI; van Oudenaarden, A. Multistability in the lactose utilization network of Escherichia coli. Nature 2004, 427, 737–740. [Google Scholar]
- Pomerening, JR; Sontag, ED; Ferrell, JE, Jr. Building a cell cycle oscillator: Hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol 2003, 5, 346–351. [Google Scholar]
- Xiong, W; Ferrell, JE, Jr. A positive-feedback-based bistable 'memory module' that governs a cell fate decision. Nature 2003, 426, 460–464. [Google Scholar]
- Bagowski, CP; Ferrell, JE. Bistability in the JNK cascade. Curr. Biol 2001, 11, 1176–1182. [Google Scholar]
- Harding, A; Tian, T; Westbury, E; Frische, E; Hancock, JF. Subcellular localization determines MAP kinase signal output. Curr. Biol 2005, 15, 869–873. [Google Scholar]
- Markevich, NI; Hoek, JB; Kholodenko, BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J. Cell Biol 2004, 164, 353–359. [Google Scholar]
- Novak, B; Tyson, JJ. Design principles of biochemical oscillators. Nat. Rev. Mol. Cell Biol 2008, 9, 981–991. [Google Scholar]
- Maithreye, R; Sarkar, RR; Parnaik, VK; Sinha, S. Delay-induced transient increase and heterogeneity in gene expression in negatively auto-regulated genecircuits. PLOS One 2008, 3, e2972. [Google Scholar]
- Coombes, S; Laing, C. Delays in activity-based neural networks. Phil. Trans. R. Soc. A 2009, 367, 1117–1129. [Google Scholar]
- Radde, N. The impact of time delays on the robustness of biological oscillators and the effect of bifurcations on the inverse problem. EURASIP J. Bioinform. Syst. Biol 2009. [Google Scholar] [CrossRef]
- Mocek, WT; Rudnicki, R; Voit, EO. Approximation of delays in biochemical systems. Math. Biosci 2005, 198, 190–216. [Google Scholar]
- Kepler, TB; Elston, TC. Stochasticity in transcriptional regulation: Origins, consequences and mathematical representations. Biophys. J 2001, 81, 3116–3136. [Google Scholar]
- Becskei, A; Kaufmann, BB; van Oudenaarden, A. Contributions of low molecule number and chromosomal positioning to stochastic gene expression. Nat. Genet 2005, 37, 937–944. [Google Scholar]
- Colman-Lerner, A; Gordon, A; Serra, E; Chin, T; Resnekov, O; Endy, D; Pesce, GC; Brent, R. Regulated cell-to-cell variation in a cell-fate decision system. Nature 2005, 437, 699–706. [Google Scholar]
- Hooshangi, S; Weiss, R. The effect of negative feedback on noise propagation in transcriptional gene networks. Chaos 2006, 16, 026108. [Google Scholar]
- Dublanche, Y; Michalodimitrakis, K; Kummerer, N; Foglierini, M; Serrano, L. Noise in transcription negative feedback loops: Simulation and experimental analysis. Mol. Syst. Biol 2006, 2, 41. [Google Scholar]
- Maithreye, R; Sinha, S. Propagation of extrinsic perturbation in a negatively auto-regulated pathway. Phys. Biol 2007, 4, 48–59. [Google Scholar]
- Levine, E; Hwa, T. Stochastic fluctuations in metabolic pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 9224–9229. [Google Scholar]
- Barkai, N; Leibler, S. Circadian clocks limited by noise. Nature 2000, 403, 267–268. [Google Scholar]
- Endy, D; Brent, R. Modelling cellular behavior. Nature 2001, 409, 391–395. [Google Scholar]
- Gonze, D; Halloy, J; Goldbeter, A. Stochastic models for circadian oscillations: Emergence of a biological rhythm. Int. J. Quantum Chem 2004, 98, 228–238. [Google Scholar]
- Goss, PGE; Peccoud, J. Quantitative modeling of stochastic systems in molecular biology by using stochastic Petri nets. Proc. Natl. Acad. Sci. USA 1998, 95, 6750–6755. [Google Scholar]
- Murphy, KF; Adams, RM; Wang, X; Balazsi, G; Collins, JJ. Tuning and controlling gene expression noise in synthetic gene networks. Nucleic Acids Res 2010, 38, 2712–2726. [Google Scholar]
- Tian, T; Burrage, K. Stochastic models for regulatory networks of the genetic toggle switch. Proc. Natl. Acad. Sci. USA 2006, 103, 8372–8377. [Google Scholar]
- Gonze, D; Jacquet, M; Goldbeter, A. Stochastic modelling of nucleocytoplasmic oscillations of the transcription factor Msn2 in yeast. J. R. Soc. Interface 2008, 5, S95–S109. [Google Scholar]
- Dunlap, JC. Molecular bases for circadian clocks. Cell 1999, 96, 271–290. [Google Scholar]
- Young, MW; Kay, SA. Time zones: A comparative genetics of circadian clocks. Nat. Rev. Genet 2001, 2, 702–715. [Google Scholar]
- Williams, JA; Sehgal, A. Molecular components of the circadian system in Drosophila. Annu. Rev. Physiol 2001, 63, 729–755. [Google Scholar]
- Reppert, SM; Weaver, DR. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol 2001, 63, 647–676. [Google Scholar]
- Barkai, N; Leibler, S. Circadian clocks limited by noise. Nature 2000, 403, 267–268. [Google Scholar]
- Goodwin, BC. Oscillatory behavior in enzymatic control processes. Adv. Enzyme Regul 1965, 3, 425–438. [Google Scholar]
- Gonze, D; Halloy, J; Goldbeter, A. Robustness of circadian rhythms with respect to molecular noise. Proc. Natl. Acad. Sci. USA 2002, 99, 673–678. [Google Scholar]
- Gillespie, DT. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem 1977, 81, 2340–2361. [Google Scholar]
- Gillespie, DT. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Phys 1976, 22, 403–434. [Google Scholar]
- Young, MW; Kay, SA. Time zones: A comparative genetics of circadian clocks. Nat Rev. Genet 2001, 2, 702–715. [Google Scholar]
- Young, MW. Life‘s 24-hour clock: Molecular control of circadian rhythms in animal cells. Trends Biochem. Sci 2001, 25, 601–606. [Google Scholar]
- Reppert, SM; Weaver, DR. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol 2001, 63, 647–676. [Google Scholar]
- Williams, JA; Sehgal, A. Molecular components of the circadian system in Drosophila. Annu. Rev. Physiol 2001, 63, 729–755. [Google Scholar]
- Preitner, N; Damiola, F; López-Molina, L; Zakany, J; Duboule, D; Albrecht, U; Schible, U. The orphan nuclear receptor REV-ERB alpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 2002, 110, 251–260. [Google Scholar]
- Leloup, JC; Goldbeter, A. Towards a detailed computational model for the mammalian circadian clock. Proc. Natl. Acad. Sci. USA 2003, 100, 7051–7056. [Google Scholar]
- Kurosawa, G; Goldbeter, A. Amplitude of circadian oscillations entrained by 24-h light-dark cycles. J. Theor. Biol 2006, 242, 478–488. [Google Scholar]
- Chabot, JR; Pedraza, JM; Luitel, P; van Oudenaarden, A. Stochastic gene expression out-of-steady- state in the cyanobacterial circadian clock. Nature 2007, 450, 1249–1252. [Google Scholar]
- Leloup, JC; Goldbeter, A. Modeling the circadian clock: From molecular mechanism to physiological disorders. BioEssays 2008, 30, 590–600. [Google Scholar]
- Goldbeter, A. Biological rhythms: Clocks for all times. Curr. Biol 2008, 18, R751–R753. [Google Scholar]
- Sprott, JC. Chaos and Time-Series Analysis; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Small, M. Applied nonlinear time series analysis: Applications in physics, physiology and finance. In Series on Nonlinear Science; World Scientific: Singapore, 2005; Series A; Volume 52. [Google Scholar]
- Yoshiharu, Y. Windhorst, U, Johansson, H, Eds.; Detection of Chaos and Fractals from Experimental Time Series. Modern Techniques in Neuroscience Research; Springer-Verlag: Berlin Heidelberg, Germany, 1999. [Google Scholar]
- Kodba, S; Perc, M; Marhl, M. Detecting chaos from a time series. Eur. J. Phys 2005, 26, 205–215. [Google Scholar]
- Perc, M. Introducing nonlinear time series analysis in undergraduate courses. Fizika A 2006, 15, 91–112. [Google Scholar]
- Aldridge, B; Haller, G; Sorger, P; Lauffenburger, D. Direct Lyapunov exponent analysis enables parametric study of transient signaling governing cell behavior. IEE Proc. Syst. Biol 2006, 153, 425–432. [Google Scholar]
- Busing, L; Schrauwen, B; Legenstein, R. Connectivity, Dynamics, and Memory in Reservoir Computing with Binary and Analog Neurons. Neural Comput 2010, 22, 1272–1311. [Google Scholar]
- Gómez-Gardeñes, J; Moreno, Y; Floría, LM. Scale-free topologies and activatory-inhibitory interactions. Chaos 2006, 16, 015114. [Google Scholar]
- Hoops, S; Sahle, S; Gauges, R; Lee, C; Pahle, J; Simus, N; Singhal, M; Xu, L; Mendes, P; Kummer, U. COPASI - a complex pathway simulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar]
- Hoppenstaedt, FC. Analysis and Simulation of Chaotic Systems; Springer-Verlag: New York, NY, USA, 2000. [Google Scholar]
- Patnaik, PR. Application of the Lyapunov exponent to detect noise-induced chaos in oscillating microbial cultures. Chaos Sol. Fractals 2005, 26, 759–765. [Google Scholar]
- Lee, YK; Shih, C; Tabeling, P; Ho, CM. Experimental study and nonlinear dynamic analysis of time-periodic micro chaotic mixers. J. Fluid Mech 2007, 575, 425–448. [Google Scholar]
- Wolf, A; Swift, JB; Swinney, HL; Vastano, JA. Determining lyapunov exponents from a time series. Phys. D 1998, 16, 285–317. [Google Scholar]
- Caccia, DC; Percival, DB; Cannon, MJ; Raymond, GM; Bassingthwaight, JB. Analyzing exact fractal time series: Evaluating dispersional analysis and rescaled range methods. Phys. A 1997, 246, 609–632. [Google Scholar]
- Raymond, GM; Percival, DB; Bassingthwaighte, JB. The spectra and periodograms of anti-correlated discrete fractional Gaussian noise. Phys. A 2003, 322, 169–179. [Google Scholar]
- Cannon, MJ; Percival, DB; Caccia, DC; Raymond, GM; Bassingthwaighte, JB. Evaluating scaled windowed variance methods for estimating the Hurst coefficient of time series. Phys. A 1997, 241, 606–626. [Google Scholar]
- Bassingthwaighte, JB; Raymond, GM. Evaluation of the dispersional analysis method for fractal time series. Ann. Biomed. Eng 1995, 23, 491–505. [Google Scholar]
- Fractal Analysis Programs of the National Simulation Resource. Available at: http://www.physiome.org/software/fractal/ (accessed on 13 September 2010).
- Eke, A; Herman, P; Kocsis, L; Kozak, LR. Fractal characterization of complexity in temporal physiological signals. Physiol. Meas 2002, 23, R1–R38. [Google Scholar]
- Goldberger, AL; Amaral, LA; Hausdorff, JM; Ivanov, PC; Peng, CK; Stanley, HE. Fractal dynamics in physiology: Alterations with disease and aging. Proc. Natl. Acad. Sci. USA 2002, 99, 2466–2472. [Google Scholar]
- Viswanathan, GM; Buldyrev, SV; Havlin, S; Stanley, HE. Quantification of DNA patchiness using long-range correlation measures. Biophys. J 1997, 72, 866–875. [Google Scholar]
- Ramanujan, VK; Biener, G; Herman, B. Scaling behavior in mitochondrial redox fluctuations. Biophys. J 2006, 90, L70–L72. [Google Scholar]
- Allegrini, P; Buiatti, M; Grigolini, P; West, BJ. Fractal brownian motion as non stationary process: An alternative paradigm for DNA sequences. Phys. Rev. E 1988, 57, 4558–4562. [Google Scholar]
- Audit, B; Vaillant, C; Arné, A; D‘Aubenton-Caraf, Y; Thermes, C. Wavelet analysis of DNA bending profiles reveals structural constraints on the evolution of genomic sequences. J. Biol. Phys 2004, 30, 33–81. [Google Scholar]
- Haimovich, AD; Byrne, B; Ramaswamy, R; Welsh, WJ. Wavelet analysis of DNA walks. J. Comput. Biol 2006, 7, 1289–1298. [Google Scholar]
- Kazachenko, VN; Astashev, ME; Grinevic, AA. Multifractal analysis of K+ channel activity. Biochemistry (Moscow) 2007, 2, 169–175. [Google Scholar]
- De la Fuente, IM; Pérez-Samartín, A; Martínez, L; García, MA; Vera-López, A. Long-range correlations in rabbit brain neural activity. Ann. Biomed. Eng 2006, 34, 295–299. [Google Scholar]
- Shannon, CE; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1997. [Google Scholar]
- Sinai, YG. On the notion of entropy of a dynamical system. Dokl. Akad. Nauk 1959, 124, 768–771. [Google Scholar]
- Ott, E. Chaos in dynamical systems; Cambridge University Press: New York, NY, USA, 1993. [Google Scholar]
- Pincus, SM. Assessing serial irregularity and its implications for health. Ann. NY. Acad. Sci 2001, 954, 245–267. [Google Scholar]
- Liu, PY; Iranmanesh, A; Keenan, DM; Pincus, SM; Veldhuis, JD. A noninvasive measure of negative-feedback strength, approximate entropy, unmasks strong diurnal variations in the regularity of LH secretion. Am. J. Physiol. Endocrinol. Metab 2007, 293, E1409–E1415. [Google Scholar]
- Roelfsema, F; Pereira, AM; Adriaanse, R; Endert, E; Fliers, E; Romijn, JA; Veldhuis, JD. Thyrotropin secretion in mild and severe primary hypothyroidism is distinguished by amplified burst mass and basal secretion with increased spikiness and approximate entropy. J. Clin. Endocrinol. Metab 2009, 95, 928–934. [Google Scholar]
- Shah, N; Evans, WS; Veldhuis, JD. Actions of estrogen on pulsatile, nyctohemeral, and entropic modes of growth hormone secretion. Am. J. Physiol 1999, 276, R1351–R1358. [Google Scholar]
- Richman, JS; Moorman, JR. Physiological time-series analysis using approximate entropy and sample entropy. Am. J. Physiol. Heart Circ. Physiol 2000, 278, H2039–H2049. [Google Scholar]
- Gu, Q; Ding, YS; Zhang, TL. Prediction of G-protein-coupled receptor classes in low homology using chou's pseudo amino acid composition with approximate entropy and hydrophobicity patterns. Protein Pept. Lett 2010, 17, 559–567. [Google Scholar]
- Blakely, NJ; Illing, L; Gauthier, JD. Controlling fast chaos in delay dynamical. Syst. Phys. Rev. Lett 2004, 92, 193901. [Google Scholar]
- Garfinkel, A; Spano, ML; Ditto, WL; Weiss, JN. Controlling cardiac chaos. Science 1992, 257, 1230–1235. [Google Scholar]
- van Wiggeren, GD; Roy, R. Communication with chaotic lasers. Science 1998, 279, 1198–1200. [Google Scholar]
- Dronov, V; Hendrey, RM; Antonsen, MT; Ott, E. Communication with a chaotic travelling wave tube microwave generator. Chaos 2004, 14, 30. [Google Scholar]
- Berridge, MJ. Berridge, Inositol triphosphate and calcium signalling. Nature 1993, 361, 315–325. [Google Scholar]
- Dixon, CJ; Cobbold, PH; Green, AK. Oscillations in cytosolic free Ca2+ induced by ADP and ATP in single rat hepatocytes display differential sensitivity to application of phorbol ester. Biochem. J 1995, 309, 145–149. [Google Scholar]
- Nielsen, K; S⊘rensen, PG; Hynne, F; Busse, HG. Sustained oscillations in glycolysis: An experimental and theoretical study of chaotic and complex periodic behavior and of quenching of simple oscillations. Biophys. Chem 1998, 72, 49–62. [Google Scholar]
- MacDonald, MJ; Fahien, LA; Buss, JD; Hasan, NM; Fallon, MJ; Kendrick, MA. Citrate oscillates in liver and pancreatic beta cell mitochondria and in INS-1 insulinoma cells. J. Biol. Chem 2003, 278, 51894–51900. [Google Scholar]
- Lebrun, P; Water, AT. Chaotic and irregular bursting electrical activity in mouse pancreatic B-cells. Biophys. J 1985, 48, 529–531. [Google Scholar]
- Ramanujan, VK; Biener, G; Herman, B. Scaling behavior in mitochondrial redox fluctuations. Biophys. J 2006, 90, L70–L72. [Google Scholar]
- Dyachok, O; Isakov, Y; Sagetorp, J; Tengholm, A. Oscillations of cyclic AMP in hormonestimulated insulin-secreting b-cells. Nature 2006, 439, 349–352. [Google Scholar]
- Dixon, CJ; Cobbold, PH; Green, AK. Oscillations in cytosolic free Ca2+ induced by ADP and ATP in single rat hepatocytes display differential sensitivity to application of phorbol ester. Biochem. J 1995, 309, 145–149. [Google Scholar]












© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez de la Fuente, I. Quantitative Analysis of Cellular Metabolic Dissipative, Self-Organized Structures. Int. J. Mol. Sci. 2010, 11, 3540-3599. https://doi.org/10.3390/ijms11093540
Martínez de la Fuente I. Quantitative Analysis of Cellular Metabolic Dissipative, Self-Organized Structures. International Journal of Molecular Sciences. 2010; 11(9):3540-3599. https://doi.org/10.3390/ijms11093540
Chicago/Turabian StyleMartínez de la Fuente, Ildefonso. 2010. "Quantitative Analysis of Cellular Metabolic Dissipative, Self-Organized Structures" International Journal of Molecular Sciences 11, no. 9: 3540-3599. https://doi.org/10.3390/ijms11093540