Construction and Characterization of a cDNA Library from Wheat Infected with Fusarium graminearum Fg 2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
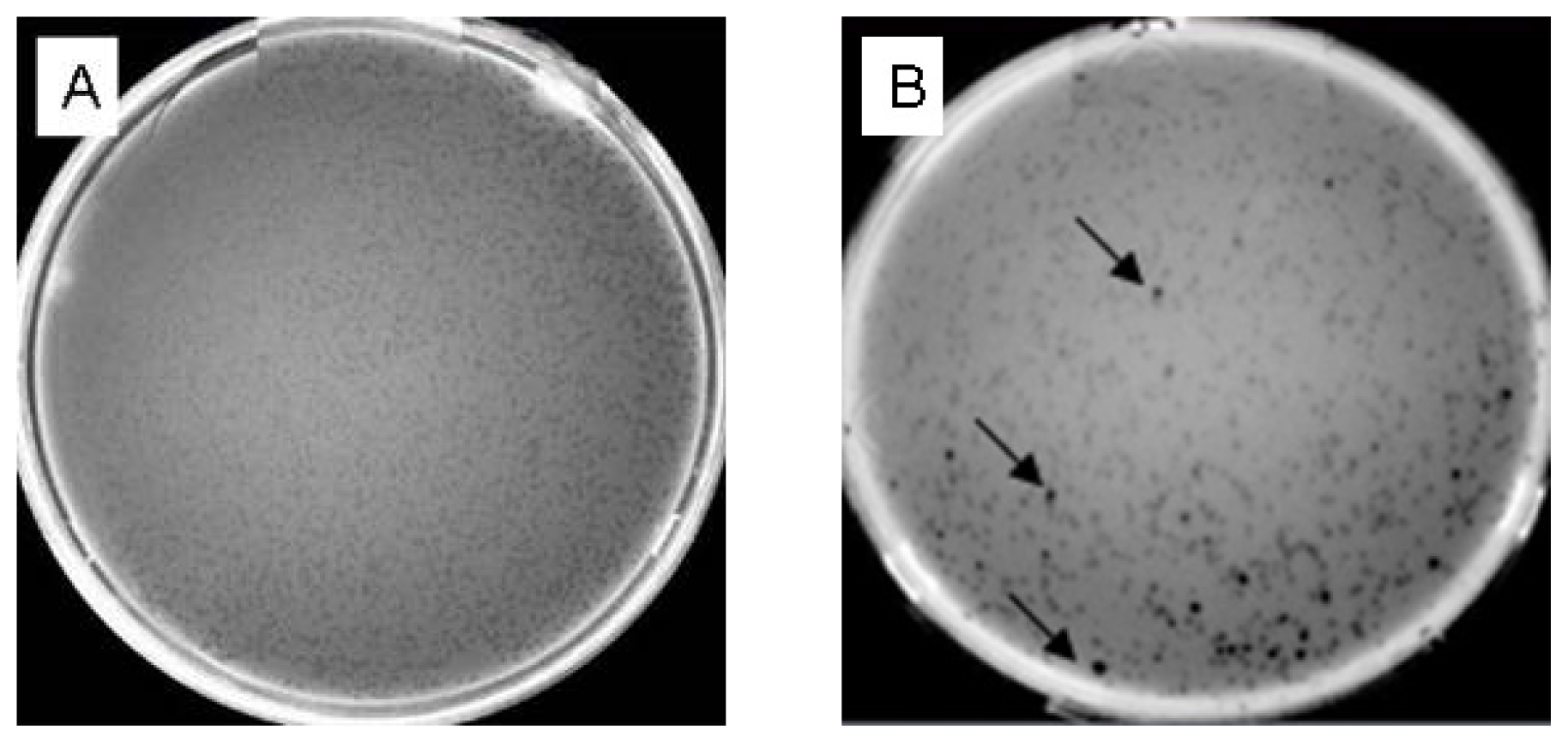{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Analysis of Total RNA
2.2. Analysis of mRNA
2.3. Analysis of Primer Extension Product
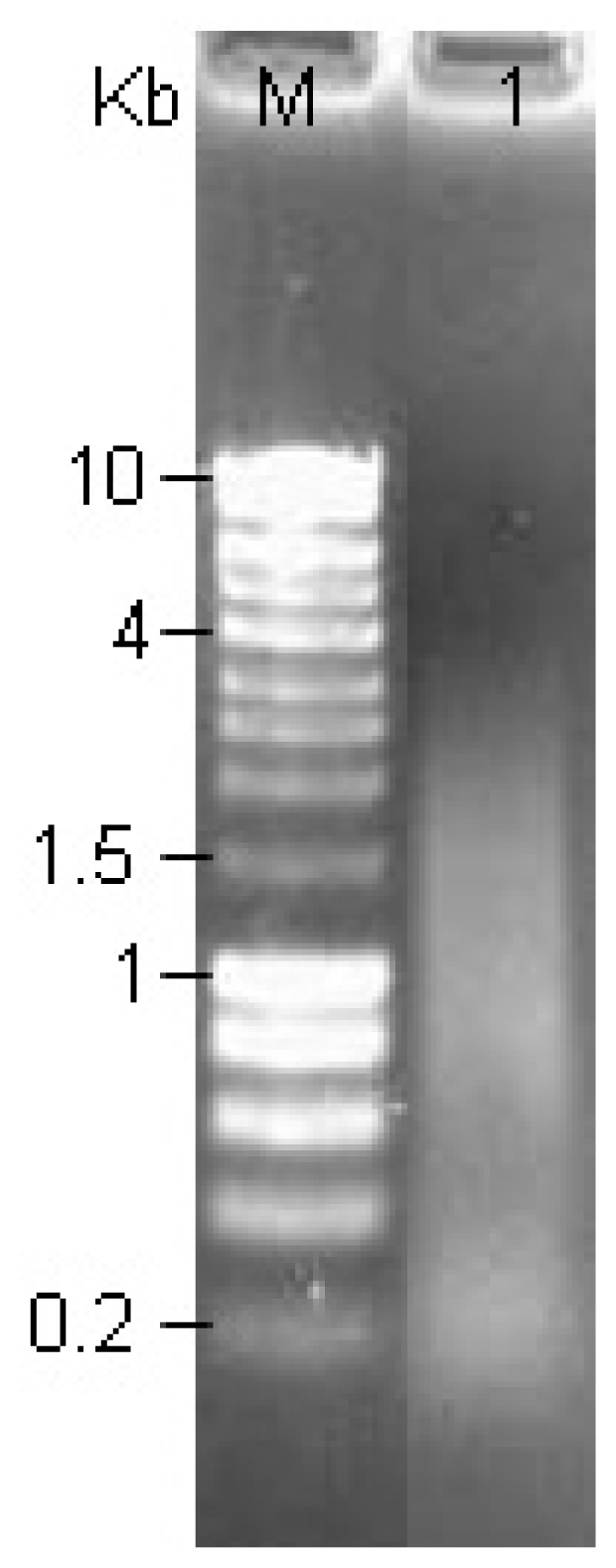
2.4. Analysis of Size-Fractionated cDNA
2.5. Analysis of the cDNA Library
2.5.1. Titering the Unamplified Library and Determining the Percentage of the Recombinants
2.5.2. Titering the Amplified Library and Determining the Percentage of the Recombinants
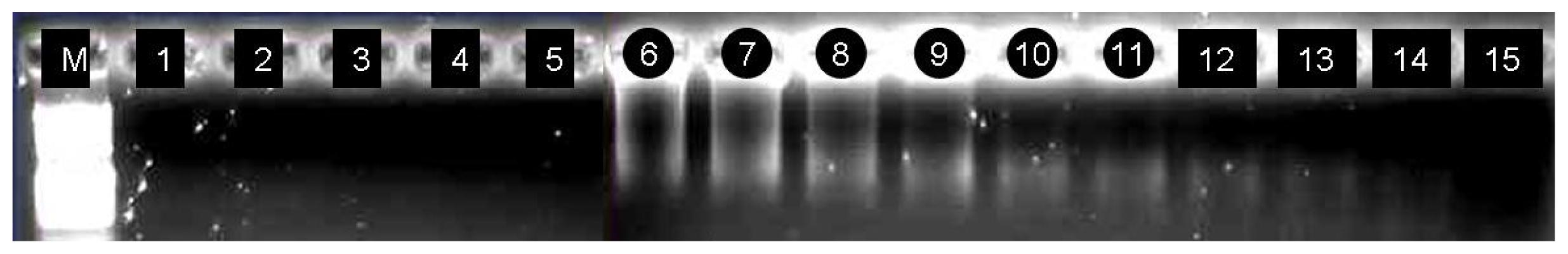
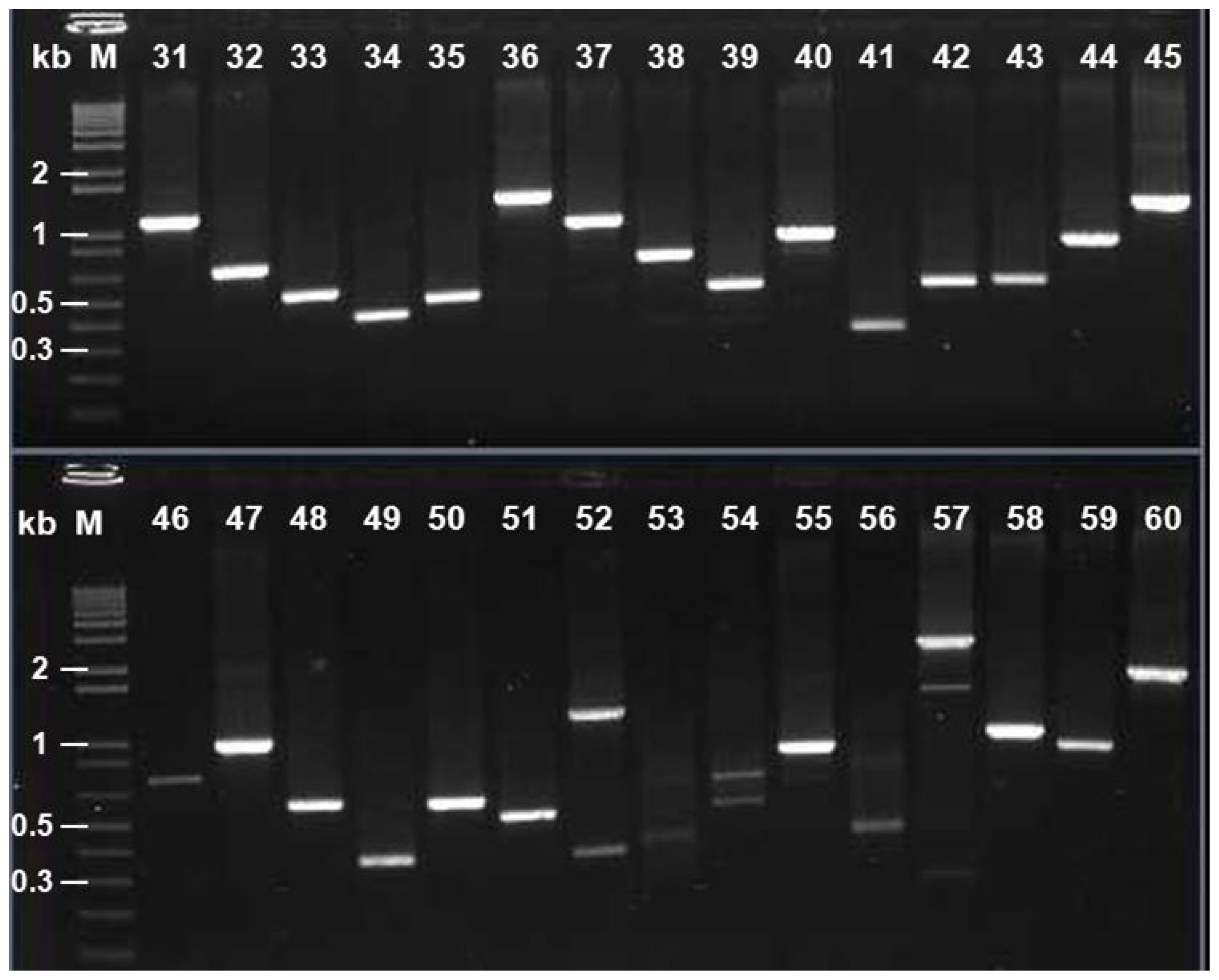
2.6. Identification of the cDNA Inserts of the Recombinants
3. Materials and Methods
3.1. Plant Materials
3.2. Pathogen and Inoculation
3.3. Extraction of Total RNA
3.4. Purification of mRNA
3.5. cDNA Synthesis
3.5.1. First-Strand
3.5.2. Amplification of cDNA by Primer Extension
3.5.3. Proteinase K Digestion
3.5.4. PCR Product Purification
3.5.5. Sfi I Digestion
3.5.6. cDNA Size Fractionation by CHROMA SPIN-400
3.6. Construction of a cDNA Library
3.6.1. Ligation of cDNA to λTriplEx2 Vector
3.6.2. λ Phage Packaging
3.6.3. Titering the Unamplified Library
3.6.4. Determining the Percentage of Recombinant Clones in Unamplified Library
3.6.5. Library Amplification
3.7. Identification of the Amplified Library
3.7.1. Titering the Amplified Library
3.7.2. Determining the Percentage of Recombinant Clones of the Amplified Library
3.7.3. Identification of the cDNA Inserts of the Recombinants
4. Conclusions
Acknowledgements
References
- Bai, GH; Shaner, G. Scab of wheat: Prospects for control. Plant Dis 1994, 78, 760–766. [Google Scholar]
- Diatchenko, L; Lar, YFC; Campbell, AP; Chenchik, A; Moqadam, F; Huang, B; Lukyanov, S; Lukyanov, K; Gurskaya, N; Sverdlov, ED; Silbert, PD. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc. Natl. Acad. Sci USA 1996, 93, 6025–6030. [Google Scholar]
- Wang, X; Feuerstein, GZ. The use of mRNA differential display for discovery of novel therapeutic targets in cardiovascular disease. Cardiovasc. Res 1997, 35, 414–421. [Google Scholar]
- Sambrook, J; Russell, DW. Molecular Cloning: A Laboratory Manual, 3rd ed; Cold Spring Harbor Lab Press: New York, NY, USA, 2002. [Google Scholar]
- Draper, MP; August, PR; Connolly, T; Packard, B; Call, KM. Efficient cloning of full-length cDNAs based on cDNA size fractionation. Genomics 2002, 79, 603–607. [Google Scholar]
- Wiemann, S; Mehrle, A; Bechtel, S; Wellenreuther, R; Pepperkok, R; Poustka, A. cDNAs for functional genomics and proteomics: The German consortium. CR Biol 2003, 326, 1003–1009. [Google Scholar]
- Castelli, V; Aury, JM; Jaillon, O; Wincker, P; Clepet, C; Menard, M; Cruaud, C; Schachter, V; Temple, G; Caboche, M; Weissenbach, J; Salanoubat, M. Whole genome sequence comparisons and “full-length” cDNA sequences: a combined approach to evaluate and improve Arabidopsis genome annotation. Genome Res 2004, 14, 406–413. [Google Scholar]
- Edery, I; Chu, LL; Sonenberg, N; Pelletier, J. An efficient strategy to isolate full-length cDNAs based on an mRNA cap retention procedure CAPture. Mol. Cell. Biol 1995, 15, 3363–3371. [Google Scholar]
- Carninci, P; Kvam, C; Kitamura, A; Ohsumi, T; Okazaki, Y; Itoh, M; Kamiya, M; Shibata, K; Sasaki, N; Izawa, M; et al. High efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics 1996, 37, 327–336. [Google Scholar]
- Frohman, MA; Dush, MK; Martin, GR. Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci USA 1988, 85, 8998–9002. [Google Scholar]
- Edwards, JB; Delort, J; Mallet, J. Oligodeoxyribonucleotide ligation to single stranded cDNAs: A new tool for cloning 5′ ends of mRNAs and for constructing cDNA libraries by in vitro amplification. Nucleic Acids Res 1991, 19, 5227–5232. [Google Scholar]
- Akowitz, A; Manuelidis, L. A novel cDNA/PCR strategy for efficient cloning of small amounts of undened RNA. Gene 1989, 81, 295–306. [Google Scholar]
- Chenchik, A; Diachenko, L; Moqadam, F; Tarabykin, V; Lukyanov, S; Siebert, PD. Full-length cDNA cloning and determination of mRNA 5′ and 3′ ends by amplification of adaptor-ligated cDNA. Biotechniques 1996, 21, 526–534. [Google Scholar]
- Fromont-Racine, M; Bertrand, E; Pictet, R; Grange, T. A highly sensitive method for mapping the 5′ termini of mRNAs. Nucleic Acids Res 1993, 21, 1683–1684. [Google Scholar]
- Sekine, S; Kato, S. Synthesis of full-length cDNA using DNA-capped mRNA. Nucleic Acids Symp 1993, 29, 143–144. [Google Scholar]
- Maruyama, K; Sugano, S. Oligo-capping: A simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides. Gene 1994, 138, 171–174. [Google Scholar]
- Efimov, V; Chakhmakhcheva, O; Archdeacon, J; Fernandez, J; Fedorkin, O; Dorokhov, Y; Atabekov, J. Detection of the 5′-cap structure of messenger RNAs with the use of the cap-jumping approach. Nucleic Acids Res 2001, 29, 4751–4759. [Google Scholar]
- Abe, S; Koyama, K; Usami, S. Construction and characterization of a vestibular-specific cDNA library using T7-based RNA amplification. J. Hum. Genet 2003, 48, 142–149. [Google Scholar]
- Maniatis, T; Hardison, RC; Lacy, E; Lauer, J; O’Connell, C; Quon, D; Sim, GK; Efstratiadis, A. The isolation of structural genes from libraries of eukaryotic DNA. Cell 1978, 15, 687–701. [Google Scholar]
- Wellenreuther, R; Schupp, I; Poustka, A; Wiemann, S. SMART amplification combined with cDNA size fractionation in order to obtain large full-length clones. BMC Genomics 2004, 5, 36. [Google Scholar]
- Zhu, YY; Machleder, EM; Chenchik, A; Li, R; Siebert, PD. Reverse transcriptase template switching: A SMART approach for full-length cDNA library construction. Biotechniques 2001, 30, 892–897. [Google Scholar]








© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Al-Taweel, K.; Fernando, W.G.D.; Brûlé-Babel, A.L. Construction and Characterization of a cDNA Library from Wheat Infected with Fusarium graminearum Fg 2. Int. J. Mol. Sci. 2011, 12, 613-626. https://doi.org/10.3390/ijms12010613
Al-Taweel K, Fernando WGD, Brûlé-Babel AL. Construction and Characterization of a cDNA Library from Wheat Infected with Fusarium graminearum Fg 2. International Journal of Molecular Sciences. 2011; 12(1):613-626. https://doi.org/10.3390/ijms12010613
Chicago/Turabian StyleAl-Taweel, Khaled, W. G. Dilantha Fernando, and Anita L. Brûlé-Babel. 2011. "Construction and Characterization of a cDNA Library from Wheat Infected with Fusarium graminearum Fg 2" International Journal of Molecular Sciences 12, no. 1: 613-626. https://doi.org/10.3390/ijms12010613