It is fast becoming standard practice in research and development to employ Design of Experiments (DOE) methods, especially in the later stages of development, when the goal shifts from screening to product and process optimization [
28]. Response Surface Methodology (RSM), such as the Central Composite Design, is the most popular class of RSM designs [
29]. In this study a Face-Centered Central Composite Design (FCCCD) (Mintab
® V15, Minitab Inc., Boston, MA, USA) was employed with two study variables namely the number of Curcumin (C) and Glyconornicotine (G) molecules. The number of molecules of curcumin (X
1) and glycosylated nornicotine (Glyconornicotine) (X
2) was selected as the independent variables studied at two levels each (2–4 for curcumin and 1–5 for Glyconornicotine). Natural variable level settings for both molecules were used. The design consisted of four cube points, five center-points in cube, four axial points (points parallel to each variable axis on a circle of radius equal to 1.0 and origin at the center-point) and 0 axial center-points. An α = 1.0 defined a geometrically square design that was both rotatable and orthogonally blocked. Orthogonally blocked designs allow for model terms and block effects to be estimated independently and minimize the variation in the regression coefficients [
30]. Rotatable designs provide the desirable property of constant prediction variance at all points that are equidistant from the design center, thus improving the quality of the prediction [
31]. Since it was not be possible to have both properties for the FCCCD design that was selected, Minitab
® opted for orthogonal blocking and simultaneously attempted to converge as close as possible to the α value for rotatability. In order to ensure the successful optimization and prediction from the design, the region of operability encompassed the region of interest. The upper and lower limits of the independent variables were determined by modeling multiple NEs simultaneously. AβP-(C)
1 and AβP-(C)
5 were eliminated due to having higher total energy values as compared to and AβP and AβP-(C)
4, respectively while AβP-(G)
6 failed to converge even after 13125 cycles (
Table 8). The number of Curcumin and Glyconornicotine molecules was in the region of interest described by the variable ranges. The design consisted of a two level full factorial with a total of 13 experimental runs.
Based on the orthogonal features of the design, a series of polynomial equations with one variable was obtained with the other six variables set at zero. Analysis of Variance (ANOVA), correlation analysis, path analysis, and regression analysis was used to analyze the dataset and statistical acceptability of the models proposed. The axial points and replicates were added to the design to provide for estimation of curvature of the models and to allow for estimation of experimental error.
2.5.1. Analysis of the Face-Centered Central Composite Design
The correlation of the independent variables and the responses were estimated by polynomial equations, using the least-square method. From a statistical point of view, three tests were used to evaluate the adequacy of the models; Student‘s
t-test which is about the significance of factors,
R-square test and Fisher tests. It was found that the individual effects were significant at a 5% significance level and only the interactions (CG), (CC), (GG) were not significant and were excluded during optimization. The test of reliability was performed by Fisher‘s variance ratio test known as the
F-test. The tabulated
F values at a 5% level of significance were between 1.08 and 12.42. Hence, it was concluded that the two variances are equal and that most of the response variation can be explained by the regression. Furthermore, the test for significance of regression confirmed that the established models provided an excellent fit to the observed data (
Figure 9). Finally, the
R2-value was found to be significantly high for the Bond Length, Bond Angle and Van der Waals forces with values of 89.9%, 89.0% and 92.8% respectively (
Table 9). These variables were considered statistically relevant for both Curcumin and Glyconornicotine and therefore considered further in this study to proceed with optimization. In general, results also revealed that the difference between the measured and the fitted values did not exceed 3% indicating that the models can adequately represent the data. At a significance level of 0.05, the mean Curcumin and Glyconornicotine appeared not to be significant to the Total Energy (
Table 9).
Estimated Regression Coefficients for all molecular attributes
where, C = curcumin and G = glyconornicotine
The sequential and adjusted sums of squares (
i.e., Seq SS and Adj SS) (
Table 3) were identical for all terms since the design matrix was orthogonal (
Table 10).
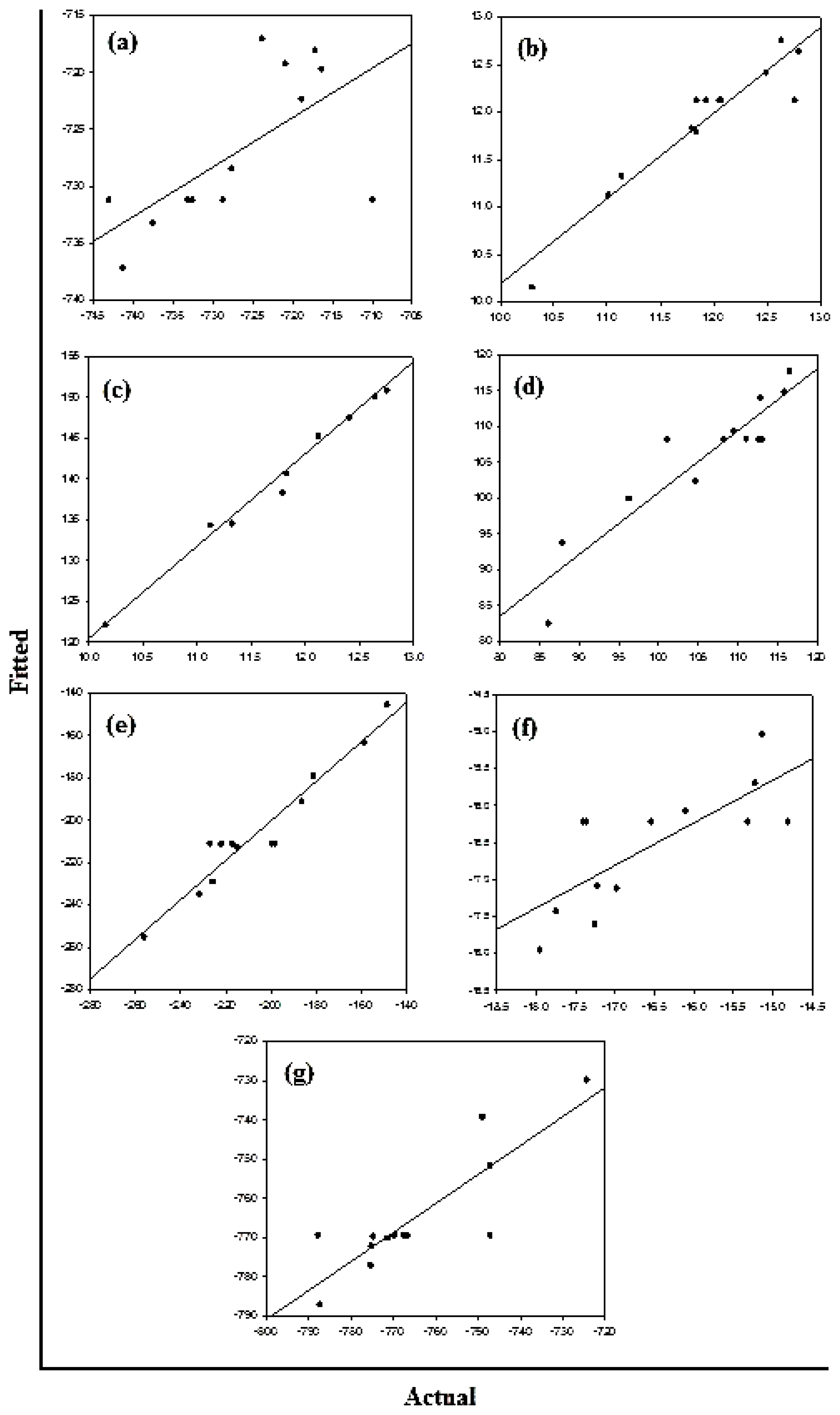
Figure 10a–c displays the diagnostic data for the design. The residuals
versus fitted profiles displayed a large randomized spread in the data points for the highest fitted values. However, it is difficult to reject the assumption of constant variance in the residuals. The residuals for the variable Van de Waals (VdW) that was selected for optimization among the Bond Length and Bond Angle followed a relatively bell-shaped curve, though the Normal probability plot had two values off linearity at either end (corresponding to high and low values). This further established the significance of VdW in binding of Curcumin and Glyconornicotine to Aβ to afford neuroprotection. However, for the variables Bond Length and Bond Angle (
Figures 10a,b) the
p-value for the Anderson Darling test for Normality was >0.05 as well as the histograms displaying a bias in the frequency of the residuals below and above baseline. Hence the null hypothesis of Normality cannot be rejected and the mean of the residuals was zero. The I-chart (Individuals control chart) in the top right hand corner of
Figures 10a–c assesses the independence assumption, and does not exhibit any concerning features. The variables Bond Length, Bond Angle and VdW for the NE combination were included in the statistical design for identifying the optimal NE combination and quantity of molecules required for neuroprotection.
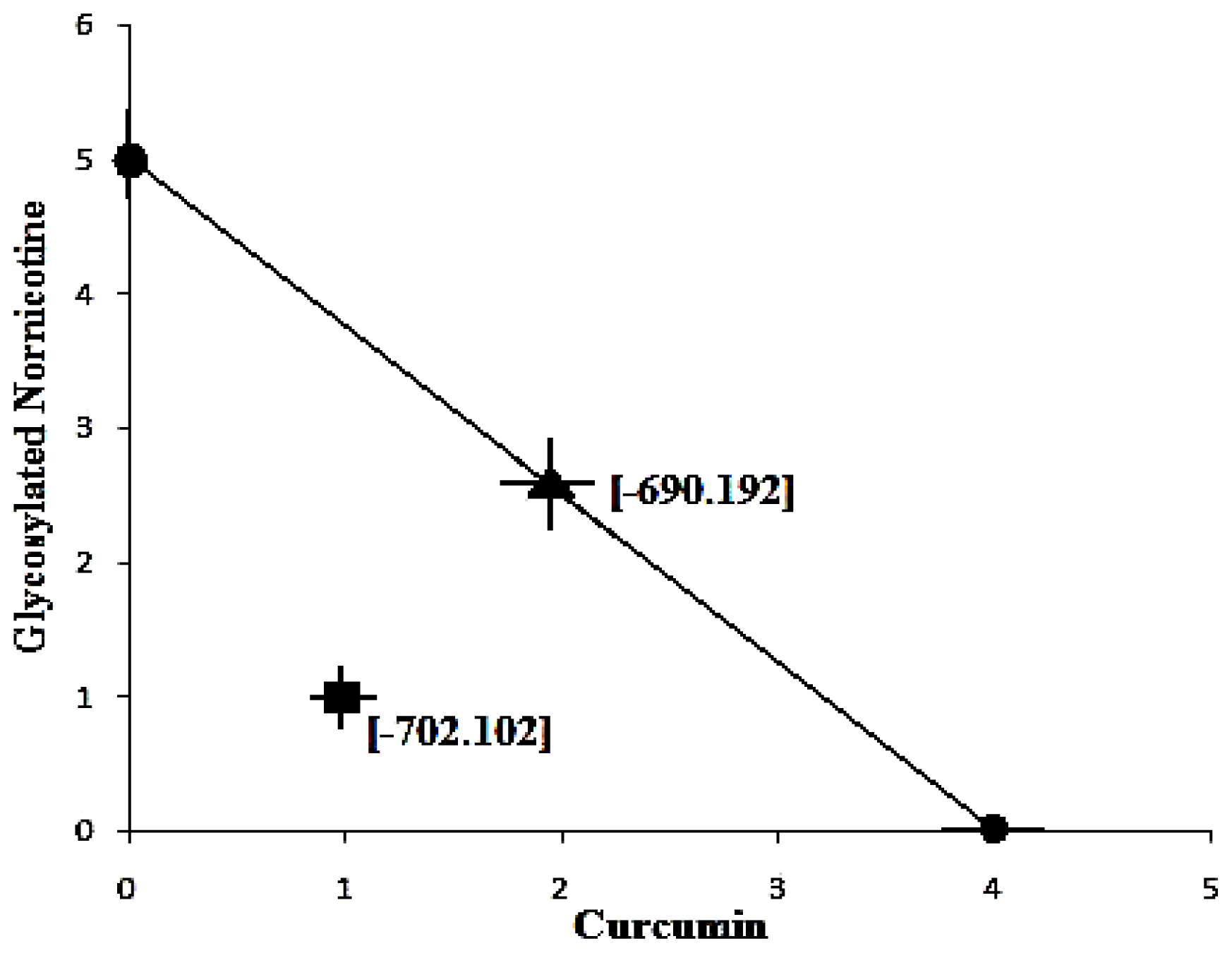
Figure 11a-c displays the 2D contour plots obtained for the three pertinent variables, Bond Length, Bond Angle and VdW employed for optimization. The optimized region (of 3 Curcumin:3 Glynornicotine) is also highlighted on the respective contour plots.
2.5.2. Response Optimization
The Response Optimizer tool of Minitab
® V15 (Minitab Inc., Boston, MA, USA) was used to obtain the optimized levels of Curcumin and Glyconornicotine based on their molecular attributes in terms of Bond Length, Bond Angle and VdW. A single optimal combination was obtained following constrained optimization of the three variables as represented in
Table 11. Upon comprehensive evaluation of feasibility searches and subsequently exhaustive grid searches, a neuroprotective combination of three molecules of Curcumin and three molecules of Glyconornicotine fulfilled the maximum requisites of an optimum combination primarily due to superior regulation of energy attributes.
Figure 12 shows the desirability plots of each constraint for the optimized combination.
Table 12 displays the local solution for Bond Length, Bond Angle and VdW in terms of the desirability score, predicted response, the actual response after Molecular Mechanics simulations of the optimized combination along with the percentage prediction errors. The prediction error for the response parameters ranged between 1.1 and 3.1% with the value of absolute error of 2.36. The low values of error indicated the high prognostic ability of the FCCCD employed in this study.

 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}