DNA Sequence Analysis of South African Helicobacter pylori Vacuolating Cytotoxin Gene (vacA)

{kind=link}
{kind=link}
Abstract
:1. Introduction
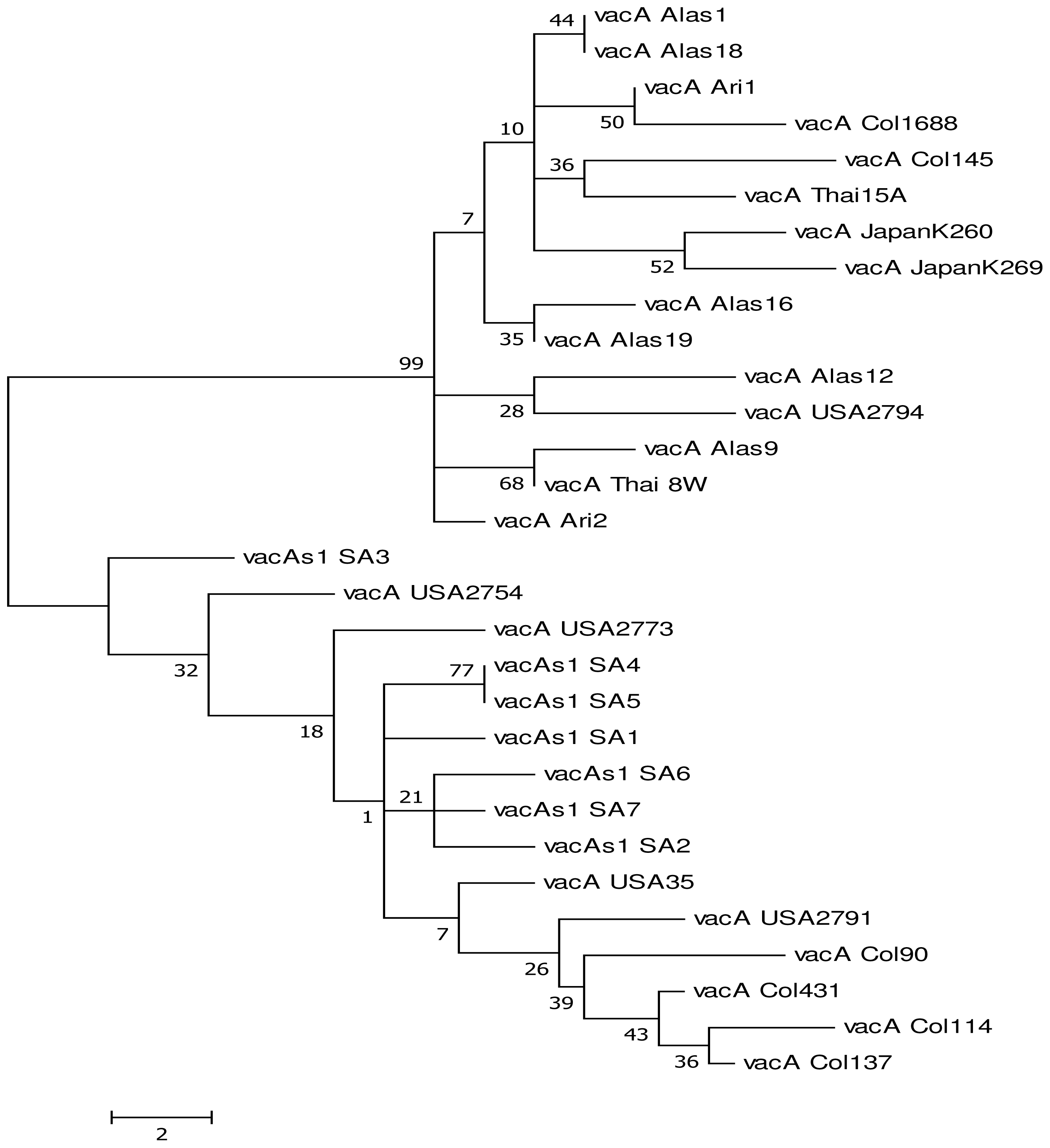
2. Results and Discussion
2.1. Results
Genetic Diversity of vacA
2.2. Discussion
3. Materials and Methods
3.1. Bacterial Strains
3.2. Molecular Characterization
3.3. Sequence Analysis
4. Conclusion
Acknowledgments
References
- Andreson, H.; Lõivukene, K.; Sillakivi, T.; Maaroos, H.-I.; Ustav, M.; Peetsalu, A.; Mikelsaa, M. Association of cagA and vacA genotypes of Helicobacter pylori with gastric diseases in Estonia. J. Clin. Microbiol 2002, 40, 298–300. [Google Scholar]
- Smith, S.I.; Kirsch, C.; Oyedeji, K.S.; Arigbabu, A.O.; Coker, A.O.; Bayerdöffer, E.; Miehlke, S. Prevalence of Helicobacter pylori vacA, cagA and iceA genotypes in Nigerian patients with duodenal ulcer disease. J. Med. Microbiol 2002, 51, 851–854. [Google Scholar]
- Matsuhisa, T.M.; Yamada, N.Y.; Kato, S.K.; Matsukura, N.M. Helicobacter pylori infection, mucosal atrophy and intestinal metaplasia in Asian populations: A comparative study in age-, gender- and endoscopic diagnosis-matched subjects. Helicobacter 2003, 8, 29–35. [Google Scholar]
- Devi, S.M.; Ahmed, I.; Khan, A.A.; Rahman, S.A.; Alvi, A.; Sechi, L.A.; Ahmed, N. Genomes of Helicobacter pylori from native Peruvians suggest a mixture of ancestral and modern lineages and reveal a western type cag-pathogenicity island. BMC Genomics 2006, 7, 191. [Google Scholar]
- Akopyanz, N.; Baranov, N.O.; Westblom, TUlf.; Kresovich, S.; Berg, D.E. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucl. Acids Res. 1992, 20, 5137–5142. [Google Scholar]
- Suerbaum, S.; Achtman, M. Population Genetics—Helicobacter pylori. In Helicobacter pylori: Physiology and Genetics; Mobley, H.L.T., Mendz, G.L., Hazell, S.L., Eds.; ASM Press: Washington, DC, USA, 2001; Volume Chapter 32. [Google Scholar]
- Kersulyte, D.; Mukhopadhyay, A.K.; Velapatiño, B.; Su, W.; Pan, Z.; Garcia, C.; Hernandez, V.; Valdez, Y.; Mistry, R.S.; Gilman, R.H.; et al. Differences in Genotypes of Helicobacter pylori from Different Human Populations. J. Bacteriol 2000, 182, 3210–3218. [Google Scholar]
- Mukhopadhyay, A.K.; Kersulyte, D.; Jeong, J.Y.; Datta, S.; Ito, Y.; Chowdhury, A.; Chowdhury, S.; Santra, A.; Bhattacharya, S.K.; Azuma, T.; Nair, G.B.; Berg, D.E. Distinctiveness of genotypes of Helicobacter pylori in Calcutta, India. J. Bacteriol 2000, 182, 3219–3227. [Google Scholar]
- Singh, V.; Mishra, S.; Maurya, P.; Rao, G.; Jain, A.K.; Dixt, V.K.; Gulati, A.K.; Nath, G. Drug resistance pattern and clonality in H. pylori. J. Infect. Dev. Ctries 2009, 3, 130–136. [Google Scholar]
- Akada, K.J.; Ogura, K.; Dailidiene, D.; Dailide, G.; Cheverud, M.J.; Berg, E.D. Helicobacter pylori tissue tropism: Mouse-colonizing strains can target different gastric niches. J. Microbiol 2003, 149, 1901–1909. [Google Scholar]
- Kidd, M.; Peek, R.M.; Lastovica, A.J.; Israel, D.A.; Kummer, A.F.; Louw, J.A. Analysis of iceA genotypes in South African Helicobacter pylori strains and relationship to clinically significant disease. Gut 2001, 49, 629–635. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Ally, R.; Mitchell, H.M.; Segal, I. Cag—A positive H. pylori aplenty in South Africa: The first systemic study of H. pylori infection in asymptomatic children in Soweto. Gut 1999, 45, A97–A98. [Google Scholar]
- Mosane, T.W.; Malope, B.I.; Ratshikhopha, M.E.; Hiss, D.C.; Sitas, F. Seroprevalence of Helicobacter pylori IgG antibodies in South African mothers and their children. Eur. J. Gastroenterol. Hepathol 2004, 16, 113–114. [Google Scholar]
- Samie, A.; Ramalivhana, J.; Igumbor, E.O.; Obi, C.L. Prevalence, haemolytic and haemagglutination activities and antibiotic susceptibility profiles of Campylobacter spp. isolated from human diarrhoeal stools in Vhembe District, South Africa. J. Health Popul. Nutr 2007, 25, 406–413. [Google Scholar]
- Dube, C.; Nkosi, T.C.; Clarke, A.M.; Mkwetshana, N.; Green, E.; Ndip, R.N. Helicobacter pylori antigenemia in an asymptomatic population of the Eastern Cape Province, South Africa: Public health implications. Rev. Environ. Health 2009, 24, 249–255. [Google Scholar]
- Tanih, N.F.; McMillan, M.; Naidoo, N.; Ndip, L.M.; Weaver, L.T.; Ndip, R.N. Prevalence of Helicobacter pylori vacA, cagA and iceA genotypes in South African patients with upper gastrointestinal diseases. Acta Trop 2010, 116, 68–73. [Google Scholar]
- Suerbaum, S.; Maynard, S.J.; Bapumia, K.; Morelli, G.; Smith, N.H.; Kunstmann, E.; Dyrek, I.; Achtman, M. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. USA 1998, 95, 12619–12624. [Google Scholar]
- Alm, R.A.; Ling, L.S.; Moir, D.T.; King, B.L.; Brown, E.D.; Doig, P.C.; Smith, D.R.; Noonan, B.; Guild, B.C.; deJonge, B.L.; et al. Genomic sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 1999, 397, 176–180. [Google Scholar]
- Alm, R.A.; Trust, J.T. Analysis of the genetic diversity of Helicobacter pylori: The tale of two genomes. J. Mol. Med 1999, 77, 834–846. [Google Scholar]
- Blaser, M.J.; Berg, D.E. Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Invest 2001, 107, 767–773. [Google Scholar]
- Maiden, M.C.J.; Malorny, B.; Achtman, M.A. Global gene pool in the Neisseriae [letter]. Mol. Microbiol 1996, 21, 1297–1298. [Google Scholar]
- Cover, L.T.; Tummuru, K.R.M.; Cao, P.; Thompson, A.S.; Blaser, J.M. Divergence of genetic sequence of the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem 1993, 269, 10566–10573. [Google Scholar]
- Wirth, T.; Wang, X.; Linz, B.; Novick, R.P.; Lum, J.K.; Blaser, M.J.; Morelli, G.; Falush, D.; Achtman, M. Distinguishing human ethnic groups via sequences from Helicobacter pylori: Lessons from Ladakh. Proc. Natl. Acad. Sci. USA 2004, 101, 4746–4751. [Google Scholar]
- Israel, D.A.; Salama, N.; Krishna, U.; Rieger, U.M.; Atherton, J.C.; Falkow, S.; Peek, R.M., Jr. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc. Natl. Acad. Sci. USA 2001, 98, 14625–14630. [Google Scholar]
- Tanih, N.F.; Okeleye, B.I.; Green, E.; Mkwetshana, N.; Clarke, A.M.; Ndip, L.M.; Ndip, R.N. Helicobacter pylori prevalence in dyspeptic patients in Eastern Cape Province: Race and disease status. South Afr. Med. J 2010, 100, 734–737. [Google Scholar]


© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tanih, N.F.; Ndip, L.M.; Ndip, R.N. DNA Sequence Analysis of South African Helicobacter pylori Vacuolating Cytotoxin Gene (vacA). Int. J. Mol. Sci. 2011, 12, 7459-7468. https://doi.org/10.3390/ijms12117459
Tanih NF, Ndip LM, Ndip RN. DNA Sequence Analysis of South African Helicobacter pylori Vacuolating Cytotoxin Gene (vacA). International Journal of Molecular Sciences. 2011; 12(11):7459-7468. https://doi.org/10.3390/ijms12117459
Chicago/Turabian StyleTanih, Nicoline F., Lucy M. Ndip, and Roland N. Ndip. 2011. "DNA Sequence Analysis of South African Helicobacter pylori Vacuolating Cytotoxin Gene (vacA)" International Journal of Molecular Sciences 12, no. 11: 7459-7468. https://doi.org/10.3390/ijms12117459