Nifedipine Protects INS-1 β-Cell from High Glucose-Induced ER Stress and Apoptosis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. MTT Assay
2.4. Hoechst 33342 Staining
2.5. Tunel Staining
2.6. Western Blot Analysis
2.7. Calcium Mobilization Assay
2.8. Insulin Staining and Glucose Stimulated Insulin Secretion (GSIS) Assay
2.9. Statistical Analysis
3. Results
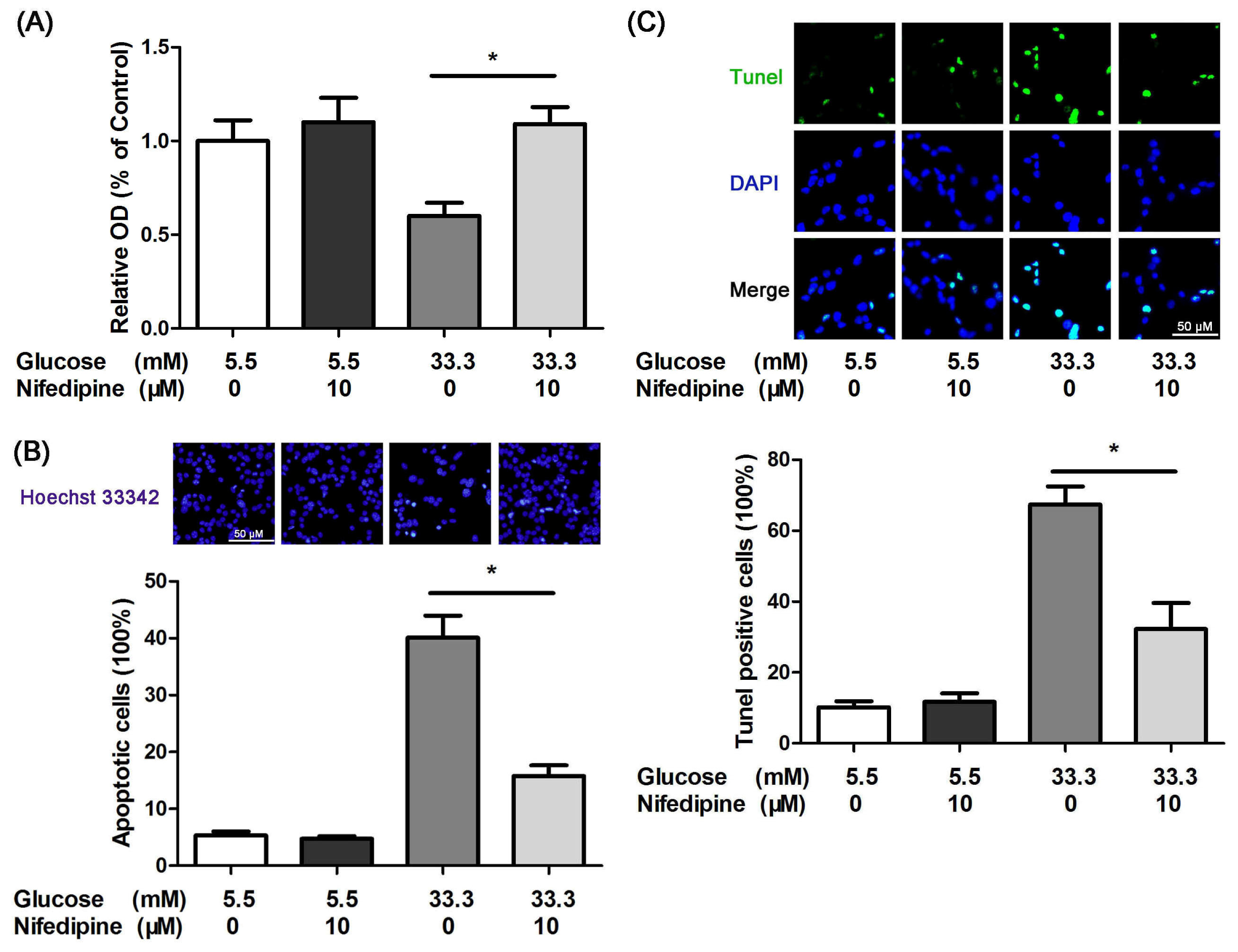
3.1. Nifedipine Protects INS-1 Cells from High Glucose-Induced Apoptosis
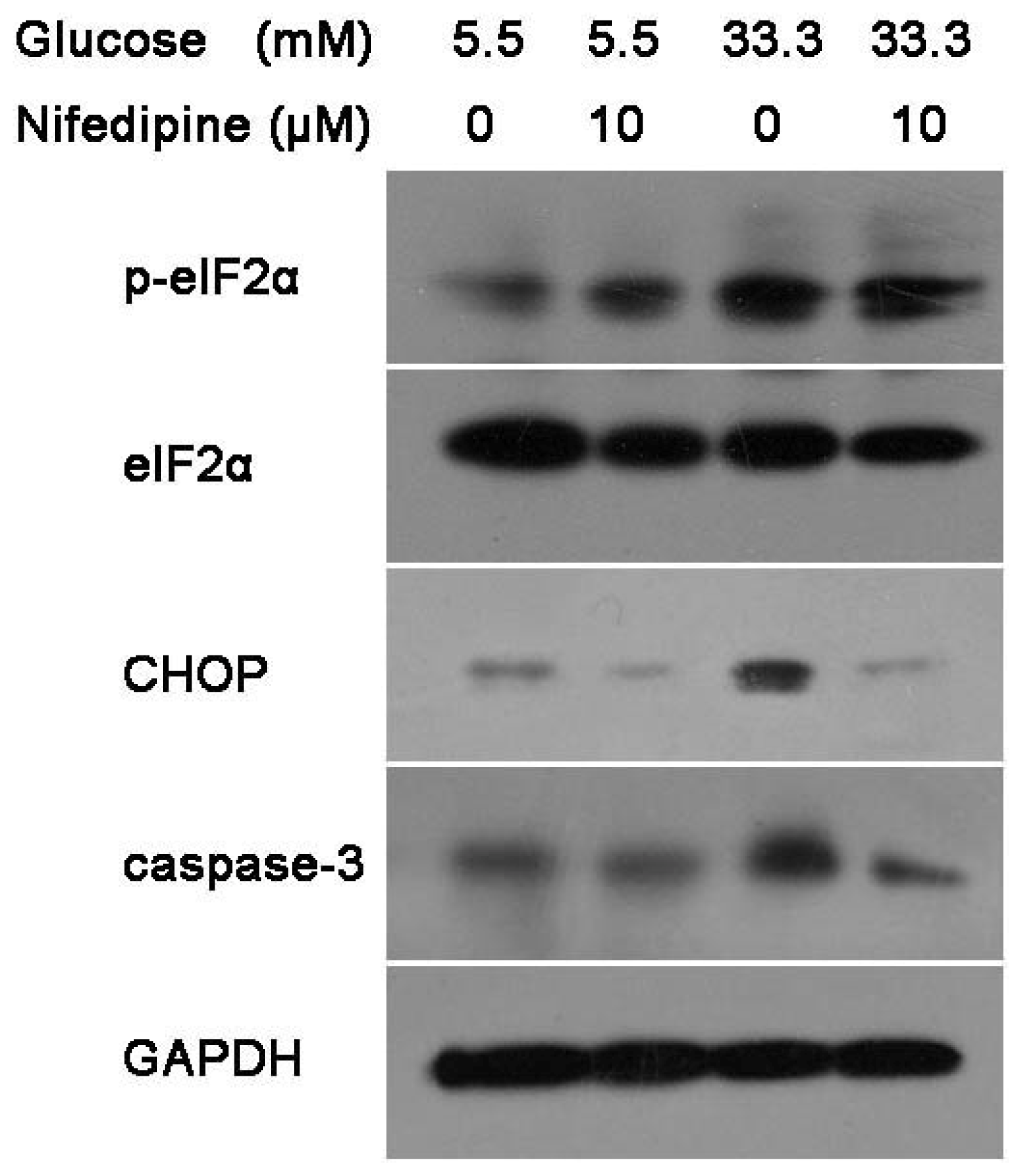
3.2. Nifedipine Prevents High Glucose-Induced β-Cell Death Through ER Stress Pathway
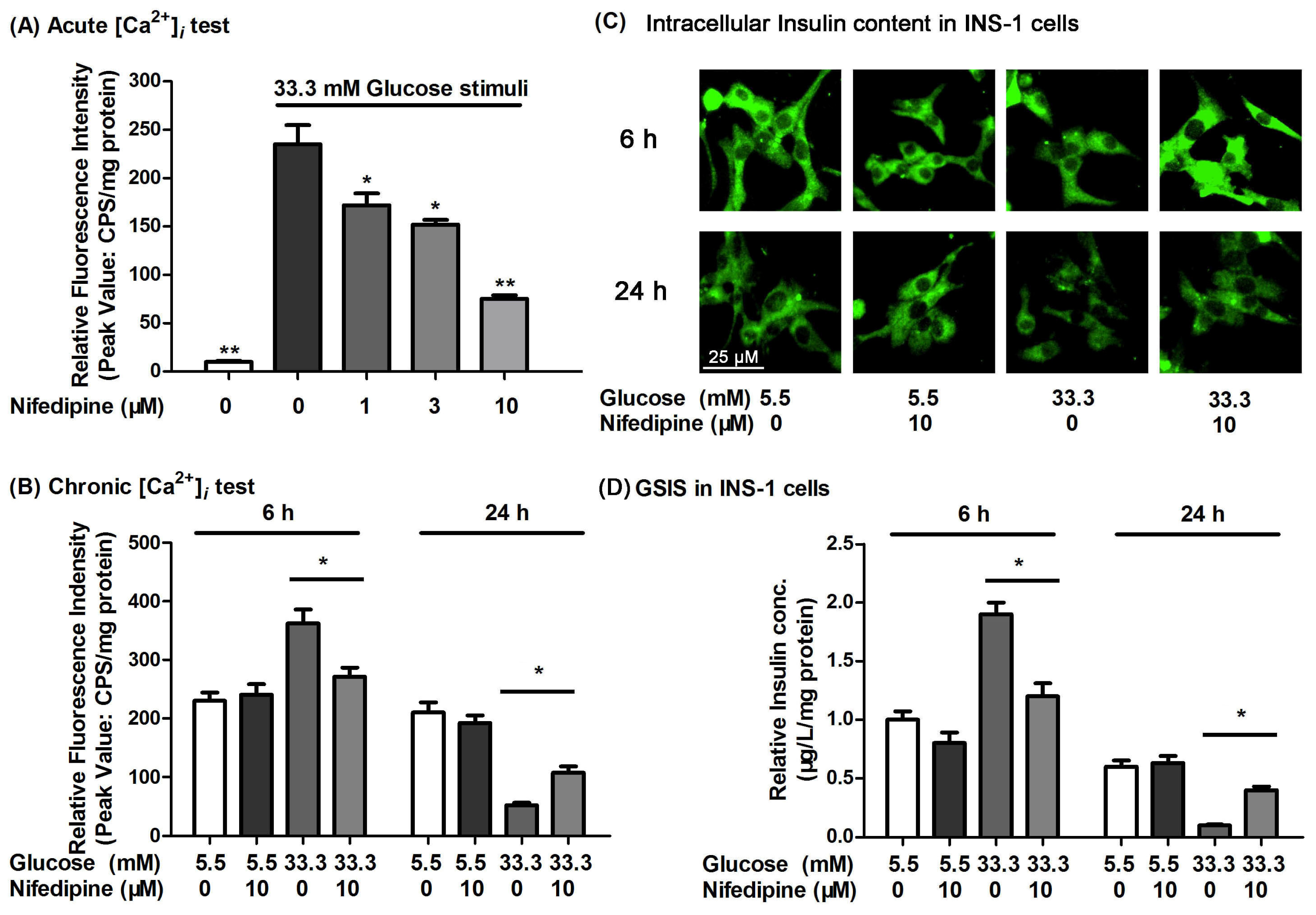
3.3. Nifedipine Improves the High Glucose-Impaired Calcium Homeostasis and GSIS in INS-1 Cells
4. Discussion
Acknowledgements
References
- Kahn, B.B. Type 2 diabetes: When insulin secretion fails to compensate for insulin resistance. Cell 1998, 92, 593–596. [Google Scholar]
- Leonardi, O.; Mints, G.; Hussain, M.A. Beta-cell apoptosis in the pathogenesis of human type 2 diabetes mellitus. Eur. J. Endocrinol 2003, 149, 99–102. [Google Scholar]
- Maedler, K.; Storling, J.; Sturis, J.; Zuellig, R.A.; Spinas, G.A.; Arkhammar, P.O.; Mandrup-Poulsen, T.; Donath, M.Y. Glucose- and interleukin-1beta-induced beta-cell apoptosis requires Ca2+ influx and extracellular signal-regulated kinase (ERK) 1/2 activation and is prevented by a sulfonylurea receptor 1/inwardly rectifying K+ channel 6.2 (SUR/Kir6.2) selective potassium channel opener in human islets. Diabetes 2004, 53, 1706–1713. [Google Scholar]
- Fei, H.; Zhao, B.; Zhao, S.; Wang, Q. Requirements of calcium fluxes and ERK kinase activation for glucose- and interleukin-1beta-induced beta-cell apoptosis. Mol. Cell. Biochem 2008, 315, 75–84. [Google Scholar]
- Maedler, K.; Spinas, G.A.; Lehmann, R.; Sergeev, P.; Weber, M.; Fontana, A.; Kaiser, N.; Donath, M.Y. Glucose induces beta-cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes 2001, 50, 1683–1690. [Google Scholar]
- Widenmaier, S.B.; Ao, Z.; Kim, S.J.; Warnock, G.; McIntosh, C.H. Suppression of p38 MAPK and JNK via Akt-mediated inhibition of apoptosis signal-regulating kinase 1 constitutes a core component of the beta-cell pro-survival effects of glucose-dependent insulinotropic polypeptide. J. Biol. Chem 2009, 284, 30372–30382. [Google Scholar]
- Zhang, Z.; Liew, C.W.; Handy, D.E.; Zhang, Y.; Leopold, J.A.; Hu, J.; Guo, L.; Kulkarni, R.N.; Loscalzo, J.; Stanton, R.C. High glucose inhibits glucose-6-phosphate dehydrogenase, leading to increased oxidative stress and beta-cell apoptosis. FASEB J 2010, 24, 1497–1505. [Google Scholar]
- Lupi, R.; Dotta, F.; Marselli, L.; del Guerra, S.; Masini, M.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar]
- Cnop, M.; Ladriere, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. Diabetes Obes. Metab 2010, 12, 76–82. [Google Scholar]
- Lupi, R.; del Prato, S. Beta-cell apoptosis in type 2 diabetes: Quantitative and functional consequences. Diabetes Metab 2008, 34, S56–S64. [Google Scholar]
- Mooradian, A.D.; Haas, M.J. Glucose-induced endoplasmic reticulum stress is independent of oxidative stress: A mechanistic explanation for the failure of antioxidant therapy in diabetes. Free Radic. Biol. Med 2011, 50, 1140–1143. [Google Scholar]
- Federici, M.; Hribal, M.; Perego, L.; Ranalli, M.; Caradonna, Z.; Perego, C.; Usellini, L.; Nano, R.; Bonini, P.; Bertuzzi, F.; et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans: A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes 2001, 50, 1290–1301. [Google Scholar]
- Dyntar, D.; Eppenberger-Eberhardt, M.; Maedler, K.; Pruschy, M.; Eppenberger, H.M.; Spinas, G.A.; Donath, M.Y. Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 2001, 50, 2105–2113. [Google Scholar]
- Diakogiannaki, E.; Morgan, N.G. Differential regulation of the ER stress response by long-chain fatty acids in the pancreatic beta-cell. Biochem. Soc. Trans 2008, 36, 959–962. [Google Scholar]
- Wang, Q.; Zhang, H.; Zhao, B.; Fei, H. IL-1beta caused pancreatic beta-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Mol. Cell. Biochem 2009, 324, 183–190. [Google Scholar]
- Harding, H.P.; Calfon, M.; Urano, F.; Novoa, I.; Ron, D. Transcriptional and translational control in the Mammalian unfolded protein response. Annu. Rev. Cell. Dev. Biol 2002, 18, 575–599. [Google Scholar]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem 2004, 279, 25935–25938. [Google Scholar]
- Thomas, S.E.; Dalton, L.E.; Daly, M.L.; Malzer, E.; Marciniak, S.J. Diabetes as a disease of endoplasmic reticulum stress. Diabetes Metab. Res. Rev 2010, 26, 611–621. [Google Scholar]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004, 11, 381–389. [Google Scholar]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Invest 2005, 115, 2656–2664. [Google Scholar]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest 2011, 121, 2118–2125. [Google Scholar]
- Ashcroft, S.J.; Ashcroft, F.M. Properties and functions of ATP-sensitive K-channels. Cell. Signal 1990, 2, 197–214. [Google Scholar]
- Chen, M.; He, H.; Zhan, S.; Krajewski, S.; Reed, J.C.; Gottlieb, R.A. Bid is cleaved by calpain to an active fragment in vitro and during myocardial ischemia/reperfusion. J. Biol. Chem 2001, 276, 30724–30728. [Google Scholar]
- Wang, H.G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar]
- Nunemaker, C.S.; Satin, L.S. Comparison of metabolic oscillations from mouse pancreatic beta cells and islets. Endocrine 2004, 25, 61–67. [Google Scholar]
- Ji, Y.; Lu, G.; Chen, G.; Huang, B.; Zhang, X.; Shen, K.; Wu, S. Microcystin-LR Induces Apoptosis via NF-kappaB/iNOS Pathway in INS-1 Cells. Int. J. Mol. Sci 2011, 12, 4722–4734. [Google Scholar]
- Meng, Z.X.; Nie, J.; Ling, J.J.; Sun, J.X.; Zhu, Y.X.; Gao, L.; Lv, J.H.; Zhu, D.Y.; Sun, Y.J.; Han, X. Activation of liver X receptors inhibits pancreatic islet beta cell proliferation through cell cycle arrest. Diabetologia 2009, 52, 125–135. [Google Scholar]
- Hu, H.; He, L.Y.; Gong, Z.; Li, N.; Lu, Y.N.; Zhai, Q.W.; Liu, H.; Jiang, H.L.; Zhu, W.L.; Wang, H.Y. A novel class of antagonists for the FFAs receptor GPR40. Biochem. Biophys. Res. Commun 2009, 390, 557–563. [Google Scholar]
- Martinez, S.C.; Tanabe, K.; Cras-Meneur, C.; Abumrad, N.A.; Bernal-Mizrachi, E.; Permutt, M.A. Inhibition of Foxo1 protects pancreatic islet beta-cells against fatty acid and endoplasmic reticulum stress-induced apoptosis. Diabetes 2008, 57, 846–859. [Google Scholar]
- Gwiazda, K.S.; Yang, T.L.; Lin, Y.; Johnson, J.D. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab 2009, 296, E690–E701. [Google Scholar]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar]
- Rizzuto, R.; Pinton, P.; Ferrari, D.; Chami, M.; Szabadkai, G.; Magalhaes, P.J.; Di Virgilio, F.; Pozzan, T. Calcium and apoptosis: Facts and hypotheses. Oncogene 2003, 22, 8619–8627. [Google Scholar]
- Schnell, S.; Schaefer, M.; Schofl, C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol. Cell. Endocrinol 2007, 263, 173–180. [Google Scholar]
- Wilding, J.P. The importance of free fatty acids in the development of Type 2 diabetes. Diabet. Med 2007, 24, 934–945. [Google Scholar]
- Ammala, C.; Eliasson, L.; Bokvist, K.; Larsson, O.; Ashcroft, F.M.; Rorsman, P. Exocytosis elicited by action potentials and voltage-clamp calcium currents in individual mouse pancreatic B-cells. J. Physiol 1993, 472, 665–688. [Google Scholar]
- Gilon, P.; Shepherd, R.M.; Henquin, J.C. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J. Biol. Chem 1993, 268, 22265–22268. [Google Scholar]
- Matsuda, Y.; Saegusa, H.; Zong, S.; Noda, T.; Tanabe, T. Mice lacking Ca(v)2.3 (alpha1E) calcium channel exhibit hyperglycemia. Biochem. Biophys. Res. Commun 2001, 289, 791–795. [Google Scholar]
- Ekinci, F.J.; Linsley, M.D.; Shea, T.B. Beta-amyloid-induced calcium influx induces apoptosis in culture by oxidative stress rather than tau phosphorylation. Brain Res. Mol. Brain Res 2000, 76, 389–395. [Google Scholar]
- Sorkin, E.M.; Clissold, S.P.; Brogden, R.N. Nifedipine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy, in ischaemic heart disease, hypertension and related cardiovascular disorders. Drugs 1985, 30, 182–274. [Google Scholar]
- Ares, M.P.; Porn-Ares, M.I.; Thyberg, J.; Juntti-Berggren, L.; Berggren, P.O.; Diczfalusy, U.; Kallin, B.; Bjorkhem, I.; Orrenius, S.; Nilsson, J. Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J. Lipid Res 1997, 38, 2049–2061. [Google Scholar]
- Sugano, M.; Tsuchida, K.; Makino, N. Nifedipine prevents apoptosis of endothelial cells induced by oxidized low-density lipoproteins. J. Cardiovasc. Pharmacol 2002, 40, 146–152. [Google Scholar]
- Orecna, M.; Hafko, R.; Toporcerova, V.; Strbak, V.; Bacova, Z. Cell swelling-induced insulin secretion from INS-1E cells is inhibited by extracellular Ca2+ and is tetanus toxin resistant. Cell. Physiol. Biochem 2010, 26, 197–208. [Google Scholar]
- Yang, S.N.; Berggren, P.O. The role of voltage-gated calcium channels in pancreatic beta-cell physiology and pathophysiology. Endocr. Rev 2006, 27, 621–676. [Google Scholar]
- Vasseur, M.; Debuyser, A.; Joffre, M. Sensitivity of pancreatic beta cell to calcium channel blockers. An electrophysiologic study of verapamil and nifedipine. Fundam. Clin. Pharmacol 1987, 1, 95–113. [Google Scholar]
- Jeffrey, K.D.; Alejandro, E.U.; Luciani, D.S.; Kalynyak, T.B.; Hu, X.; Li, H.; Lin, Y.; Townsend, R.R.; Polonsky, K.S.; Johnson, J.D. Carboxypeptidase E mediates palmitate-induced beta-cell ER stress and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 8452–8457. [Google Scholar]
- Choi, S.E.; Kim, H.E.; Shin, H.C.; Jang, H.J.; Lee, K.W.; Kim, Y.; Kang, S.S.; Chun, J.; Kang, Y. Involvement of Ca2+-mediated apoptotic signals in palmitate-induced MIN6N8a beta cell death. Mol. Cell. Endocrinol 2007, 272, 50–62. [Google Scholar]
- Navarro-Tableros, V.; Fiordelisio, T.; Hernandez-Cruz, A.; Hiriart, M. Physiological development of insulin secretion, calcium channels, and GLUT2 expression of pancreatic rat beta-cells. Am. J. Physiol. Endocrinol. Metab 2007, 292, E1018–E1029. [Google Scholar]
- Nolan, C.J.; Madiraju, M.S.; Delghingaro-Augusto, V.; Peyot, M.L.; Prentki, M. Fatty acid signaling in the beta-cell and insulin secretion. Diabetes 2006, 55, S16–S23. [Google Scholar]
- Tamareille, S.; Mignen, O.; Capiod, T.; Rucker-Martin, C.; Feuvray, D. High glucose-induced apoptosis through store-operated calcium entry and calcineurin in human umbilical vein endothelial cells. Cell Calcium 2006, 39, 47–55. [Google Scholar]
- Rudijanto, A. Calcium channel blocker (diltiazem) inhibits apoptosis of vascular smooth muscle cell exposed to high glucose concentration through lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) pathway. Acta Med. Indones 2010, 42, 59–65. [Google Scholar]
- Prentki, M.; Nolan, C.J. Islet beta cell failure in type 2 diabetes. J. Clin. Invest 2006, 116, 1802–1812. [Google Scholar]
- Shafrir, E. Development and consequences of insulin resistance: Lessons from animals with hyperinsulinaemia. Diabetes Metab 1996, 22, 122–131. [Google Scholar]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar]
- Taylor, J.T.; Huang, L.; Keyser, B.M.; Zhuang, H.; Clarkson, C.W.; Li, M. Role of high-voltage-activated calcium channels in glucose-regulated beta-cell calcium homeostasis and insulin release. Am. J. Physiol. Endocrinol. Metab 2005, 289, E900–E908. [Google Scholar]
- Yang, M.; Chisholm, J.W.; Soelaiman, S.; Shryock, J.C. Sulfonylureas uncouple glucose-dependence for GPR40-mediated enhancement of insulin secretion from INS-1E cells. Mol. Cell. Endocrinol 2010, 315, 308–313. [Google Scholar]
- Yang, J.; Robert, C.E.; Burkhardt, B.R.; Young, R.A.; Wu, J.; Gao, Z.; Wolf, B.A. Mechanisms of glucose-induced secretion of pancreatic-derived factor (PANDER or FAM3B) in pancreatic beta-cells. Diabetes 2005, 54, 3217–3228. [Google Scholar]



© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, Y.; Gao, L.; Li, Y.; Chen, H.; Sun, Z. Nifedipine Protects INS-1 β-Cell from High Glucose-Induced ER Stress and Apoptosis. Int. J. Mol. Sci. 2011, 12, 7569-7580. https://doi.org/10.3390/ijms12117569
Wang Y, Gao L, Li Y, Chen H, Sun Z. Nifedipine Protects INS-1 β-Cell from High Glucose-Induced ER Stress and Apoptosis. International Journal of Molecular Sciences. 2011; 12(11):7569-7580. https://doi.org/10.3390/ijms12117569
Chicago/Turabian StyleWang, Yao, Lu Gao, Yuan Li, Hong Chen, and Zilin Sun. 2011. "Nifedipine Protects INS-1 β-Cell from High Glucose-Induced ER Stress and Apoptosis" International Journal of Molecular Sciences 12, no. 11: 7569-7580. https://doi.org/10.3390/ijms12117569