Evaluation of Internal Reference Genes for Quantitative Expression Analysis by Real-Time PCR in Ovine Whole Blood

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Expression Level of Candidate Reference Genes
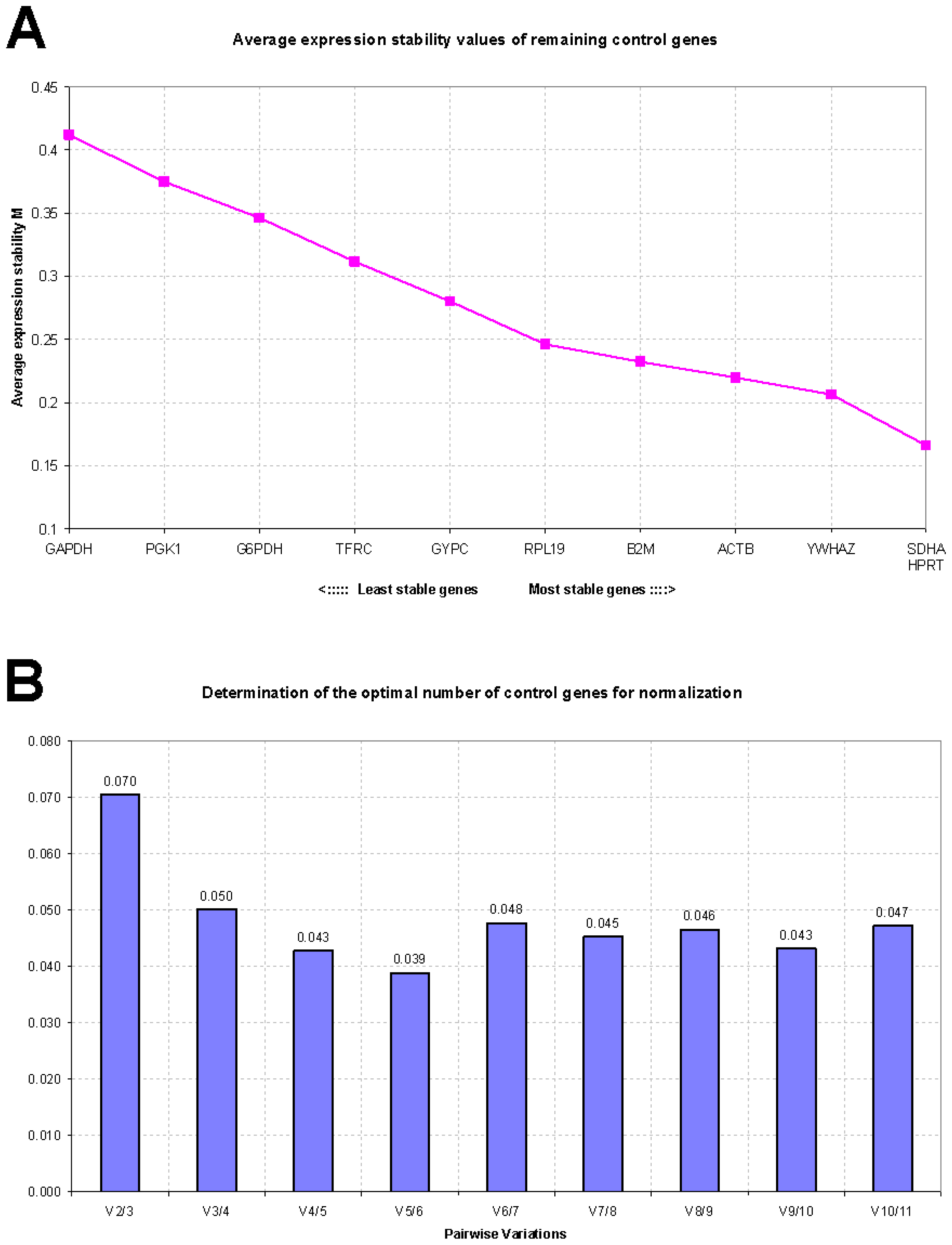
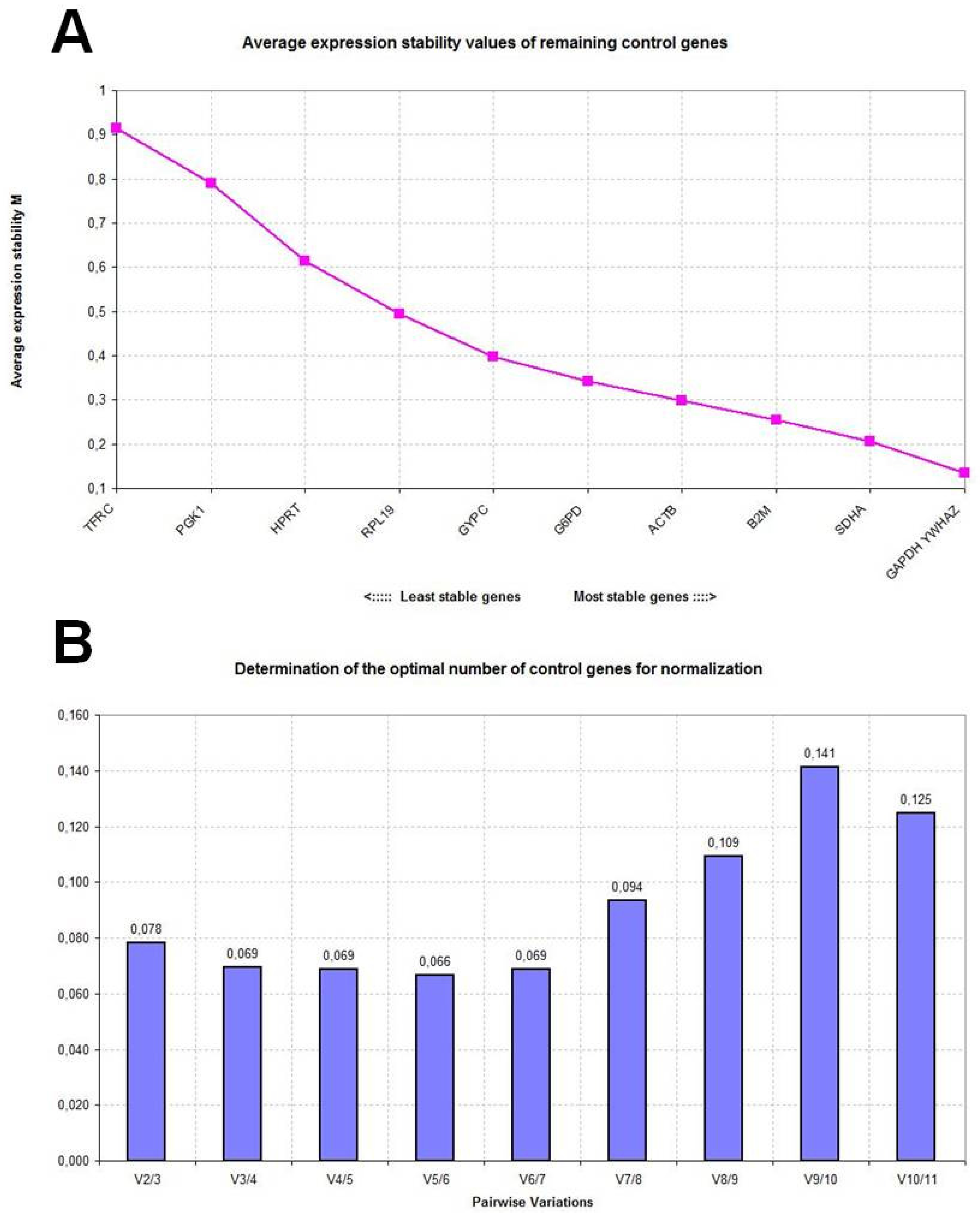
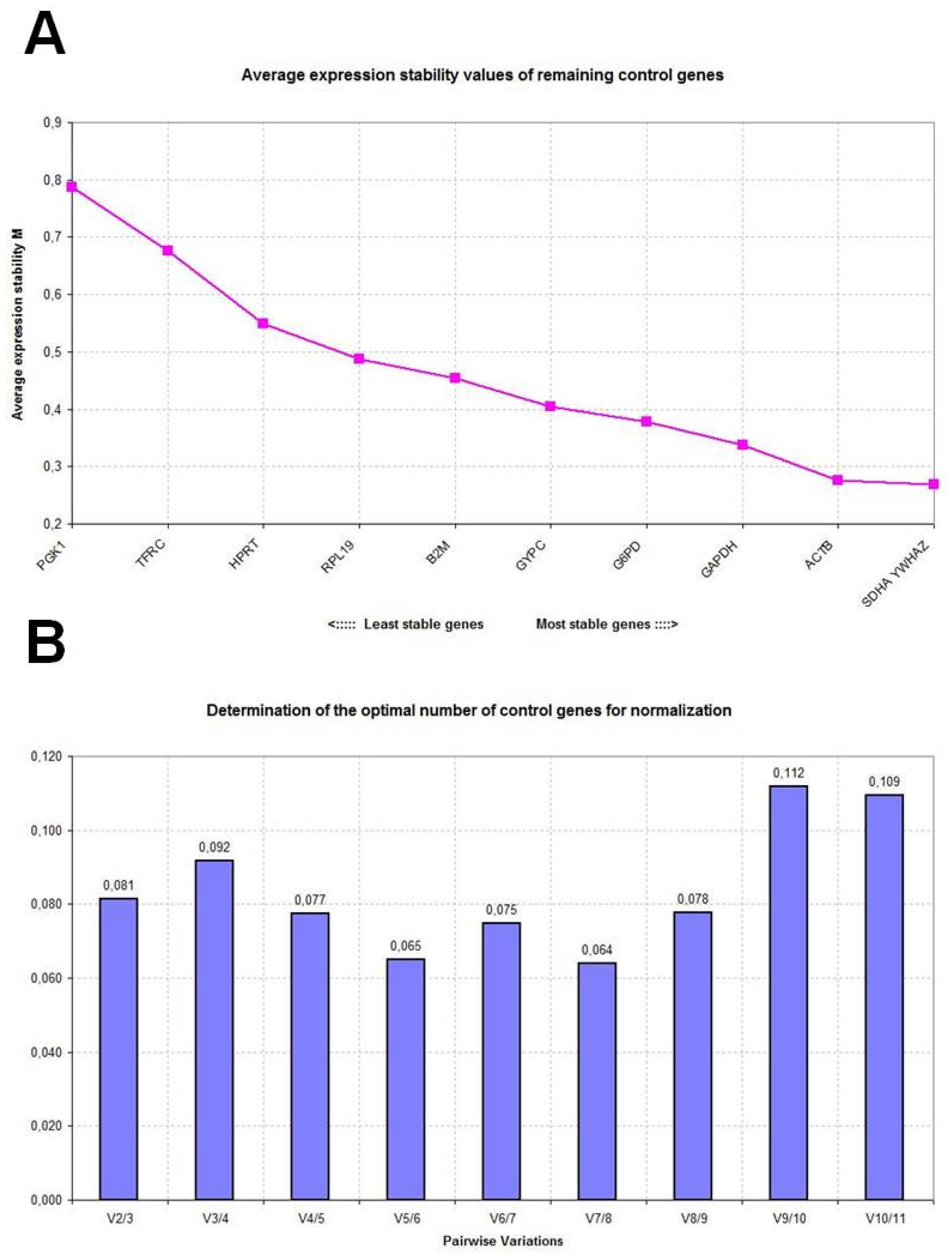
2.2. GeNorm Analysis
2.3. NormFinder Analysis
2.4. Evaluation of the Analysed Reference Genes
3. Experimental Section
3.1. Sample Collection, Nucleic Acid Extraction and cDNA Synthesis
3.2. Selection of Genes and qRT-PCR Primer Design
3.3. Quantitative RT-PCR
3.4. Data Analysis
4. Conclusions
Acknowledgments
References
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002, 3. [Google Scholar] [CrossRef] [Green Version]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 2005, 6, 279–284. [Google Scholar]
- Selvey, S.; Thompson, E.W.; Matthaei, K.; Lea, R.A.; Irving, M.G.; Griffiths, L.R. β-actin—an unsuitable internal control for RT-PCR. Mol. Cell. Probes 2001, 15, 307–311. [Google Scholar]
- Ohl, F.; Jung, M.; Xu, C.; Stephan, C.; Rabien, A.; Burkhardt, M.; Nitsche, A.; Kristiansen, G.; Loening, S.A.; Radonić, A.; Jung, K. Gene expression studies in prostate cancer tissue: Which reference gene should be selected for normalization? J. Mol. Med 2005, 83, 1014–1024. [Google Scholar]
- Garcia-Crespo, D.; Juste, R.A.; Hurtado, A. Selection of ovine housekeeping genes for normalisation by real-time RT-PCR; analysis of PrP gene expression and genetic susceptibility to scrapie. BMC Vet. Res 2005, 1, 3. [Google Scholar]
- Lampo, E.; Van Poucke, M.; Vandesompele, J.; Erkens, T.; Van Zeveren, A.; Peelman, L.J. Positive correlation between relative mRNA expression of PRNP and SPRN in cerebral and cerebellar cortex of sheep. Mol. Cell. Probes 2009, 23, 60–64. [Google Scholar]
- Passmore, M.; Nataatmadja, M.; Fraser, J.F. Selection of reference genes for normalisation of real-time RT-PCR in brain-stem death injury in Ovis aries. BMC Mol. Biol 2009, 10, 72. [Google Scholar]
- Lyahyai, J.; Serrano, C.; Ranera, B.; Badiola, J.J.; Zaragoza, P.; Martin-Burriel, I. Effect of scrapie on the stability of housekeeping genes. Anim. Biotechnol 2010, 21, 1–13. [Google Scholar]
- Taylor, D.L.; Zhong, L.; Begg, D.J.; de Silva, K.; Whittington, R.J. Toll-like receptor genes are differentially expressed at the sites of infection during the progression of Johne’s disease in outbred sheep. Vet. Immunol. Immunopathol 2008, 124, 132–151. [Google Scholar]
- Miele, G.; Manson, J.; Clinton, M. A novel erythroid-specific marker of transmissible spongiform encephalopathies. Nat Med 2001, 7, 361–364. [Google Scholar]
- Brown, A.R.; Blanco, A.R.; Miele, G.; Hawkins, S.A.; Hopkins, J.; Fazakerley, J.K.; Manson, J.; Clinton, M. Differential expression of erythroid genes in prion disease. Biochem. Biophys. Res. Commun 2007, 364, 366–371. [Google Scholar]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol 1999, 75, 291–295. [Google Scholar]
- Goidin, D.; Mamessier, A.; Staquet, M.J.; Schmitt, D.; Berthier-Vergnes, O. Ribosomal 18S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and β-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal. Biochem 2001, 295, 17–21. [Google Scholar]
- Dheda, K.; Huggett, J.F.; Bustin, S.A.; Johnson, M.A.; Rook, G.; Zumla, A. Validation of housekeeping genes for normalizing RNA expression in real-time PCR. Biotechniques 2004, 37. [Google Scholar]
- Jemiolo, B.; Trappe, S. Single muscle fiber gene expression in human skeletal muscle: Validation of internal control with exercise. Biochem. Biophys. Res. Commun 2006, 320, 1043–1050. [Google Scholar]
- Andersen, C.L.; Ledet-Jensen, J.; Ørntoft, T. Normalization of real-time quantitative RT-PCR data: a model based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004, 64, 5245–5250. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 2009, 55, 611–622. [Google Scholar]
- De Risi, J.L.; Iyer, V.R.; Brown, P.O. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 1997, 278, 680–686. [Google Scholar]
- Anstaett, O.L.; Brownlie, J.; Collins, M.E.; Thomas, C.J. Validation of endogenous reference genes for RT-qPCR normalisation in bovine lymphoid cells (BL-3) infected with Bovine Viral Diarrhoea Virus (BVDV). Vet. Immunol. Immunopathol 2010, 137, 201–207. [Google Scholar]
- Mehta, R.; Birerdinc, A.; Hossain, N.; Afendy, A.; Chandhoke, V.; Younossi, Z.; Baranova, A. Validation of endogenous reference genes for qRT-PCR analysis of human visceral adipose samples. BMC Mol. Biol 2010, 11, 39. [Google Scholar]
- Piehler, A.P.; Grimholt, R.M.; Ovstebø, R.; Berg, J.P. Gene expression results in lipopolysaccharide-stimulated monocytes depend significantly on the choice of reference genes. BMC Immunol 2010, 11, 21. [Google Scholar]
- Spalenza, V.; Girolami, F.; Bevilacqua, C.; Riondato, F.; Rasero, R.; Nebbia, C.; Sacchi, P.; Martin, P. Identification of internal control genes for quantitative expression analysis by real-time PCR in bovine peripheral lymphocytes. Vet J 2010. [Google Scholar] [CrossRef]
- Stamova, B.S.; Apperson, M.; Walker, W.L.; Tian, Y.; Xu, H.; Adamczy, P.; Zhan, X.; Liu, D.; Ander, B.P.; Liao, I.H.; et al. Identification and validation of suitable endogenous reference genes for gene expression studies in human peripheral blood. BMC Med. Genomics 2009, 2, 49. [Google Scholar]
- Le Van Kim, C.; Colin, Y.; Mitjavila, M.T.; Clerget, M.; Dubart, A.; Nakazawa, M.; Vainchenker, W.; Cartron, J.P. Structure of the promoter region and tissue specificity of the human glycophorin C gene. J. Biol. Chem 1989, 264, 20407–20414. [Google Scholar]
- Olsvik, P.A.; Lie, K.K.; Jordal, A.E.; Nilsen, T.O.; Hordvik, I. Evaluation of potential reference genes in real-time RT-PCR studies of Atlantic salmon. BMC Mol. Biol 2005, 6, 21. [Google Scholar]
- Brattelid, T.; Winer, L.H.; Levy, F.O.; Liestøl, K.; Sejersted, O.M.; Andersson, K.B. Reference gene alternatives to GAPDH in rodent and human heart failure gene expression studies. BMC Mol. Biol 2010, 11, 22. [Google Scholar]
- RTPrimerDB Website. Available online: http://www.rtprimerdb.org accessed on 22 July 2011.
- Pattyn, F.; Speleman, F.; De Paepe, A.; Vandesompele, J. RTPrimerDB: The Real-Time PCR primer and probe database. Nucleic Acids Res 2003, 31, 122–123. [Google Scholar]
- Pattyn, F.; Robbrecht, P.; Speleman, F.; De Paepe, A.; Vandesompele, J. RTPrimerDB: The Real-Time PCR primer and probe database, major update 2006. Nucleic Acids Res 2006, 34, D684–D688. [Google Scholar]
- Lefever, S.; Vandesompele, J.; Speleman, F.; Pattyn, F. RTPrimerDB: The portal for real-time PCR primers and probes. Nucleic Acids Res 2008, 37, D942–D945. [Google Scholar]
- Schroeder, A.; Mueller, O.; Stocker, S.; Salowsky, R.; Leiber, M.; Gassmann, M.; Lightfoot, S.; Menzel, W.; Granzow, M.; Ragg, T. The RIN: An RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol 2006, 7, 3. [Google Scholar]
- Primer3 (v. 0.4.0). Available online: http://frodo.wi.mit.edu/primer3 accessed on 22 July 2011.
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007, 8, R19. [Google Scholar]
- Radonić, A.; Thulke, S.; Bae, H.G.; Müller, M.A.; Siegert, W.; Nitsche, A. Reference gene selection for quantitative real-time PCR analysis in virus infected cells: SARS corona virus, Yellow fever virus, Human Herpesvirus-6, Camelpox virus and Cytomegalovirus infections. Virol. J 2005, 2, 7. [Google Scholar]
- Spinsanti, G.; Panti, C.; Lazzeri, E.; Marsili, L.; Casini, S.; Frati, F.; Fossi, C.M. Selection of reference genes for quantitative RT-PCR studies in striped dolphin (Stenella coeruleoalba) skin biopsies. BMC Mol. Biol 2006, 7, 32. [Google Scholar]
- Nygard, A.B.; Jørgensen, C.B.; Cirera, S.; Fredholm, M. Selection of reference genes for gene expression studies in pig tissues using SYBR green qPCR. BMC Mol. Biol 2007, 8, 67. [Google Scholar]
- Cappelli, K.; Felicetti, M.; Capomaccio, S.; Spinanti, G.; Silvestrelli, M.; Supplizi, A.V. Exercise induced stress in horses: selection of the most stable reference genes for quantitative RT-PCR normalization. BMC Mol. Biol 2008, 9, 49. [Google Scholar]
- Schlotter, Y.M.; Veenhof, E.Z.; Brinkhof, B.; Rutten, V.P.; Spee, B.; Willemse, T.; Penning, L.C. A GeNorm algorithm-based selection of reference genes for quantitative real-time PCR in skin biopsies of healthy dogs and dogs with atopic dermatitis. Vet. Immunol. Immunopathol 2009, 129, 115–118. [Google Scholar]
- Kadarmideen, H.N.; von Rohr, P.; Janss, L.L. From genetical genomics to systems genetics: Potential applications in quantitative genomics and animal breeding. Mamm. Genome 2006, 17, 548–564. [Google Scholar]
- Hu, X.; Gao, Y.; Feng, C.; Liu, Q.; Wang, X.; Du, Z.; Wang, Q.; Li, N. Advanced technologies for genomic analysis in farm animals and its application for QTL mapping. Genetica 2009, 136, 371–386. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Control sheep (n = 18) | Disease-stressed sheep (n = 10) | Combined groups (n = 10 + 10) | |||
|---|---|---|---|---|---|
| Gene symbol | Stability value (M) | Gene symbol | Stability value (M) | Gene symbol | Stability value (M) |
| SDHA | 0.316 | YWHAZ | 0.624 | SDHA | 0.591 |
| YWHAZ | 0.342 | GAPDH | 0.634 | YWHAZ | 0.593 |
| HPRT | 0.344 | SDHA | 0.667 | ACTB | 0.596 |
| RPL19 | 0.361 | ACTB | 0.701 | G6PD | 0.666 |
| B2M | 0.362 | B2M | 0.720 | GAPDH | 0.681 |
| ACTB | 0.377 | G6PD | 0.740 | GYPC | 0.698 |
| GYPC | 0.412 | GYPC | 0.848 | RPL19 | 0.746 |
| TFRC | 0.455 | RPL19 | 1.000 | B2M | 0.760 |
| G6PD | 0.486 | HPRT | 1.188 | HPRT | 0.882 |
| PGK1 | 0.498 | PGK1 | 1.466 | TFRC | 1.158 |
| GAPDH | 0.579 | TFRC | 1.485 | PGK1 | 1.295 |
| Control sheep (n = 18) | Disease-stressed sheep (n = 10) | Combined groups (n = 10 + 10) | |||
|---|---|---|---|---|---|
| Gene symbol | Stability value (ρ) | Gene symbol | Stability value (ρ) | Gene symbol | Stability value (ρ) |
| SDHA | 0.068 | YWHAZ | 0.046 | SDHA | 0.093 |
| YWHAZ | 0.125 | GAPDH | 0.064 | YWHAZ | 0.096 |
| HPRT | 0.132 | SDHA | 0.188 | ACTB | 0.099 |
| B2M | 0.146 | ACTB | 0.220 | GAPDH | 0.167 |
| RPL19 | 0.155 | B2M | 0.229 | RPL19 | 0.175 |
| ACTB | 0.174 | G6PD | 0.248 | G6PD | 0.184 |
| GYPC | 0.201 | GYPC | 0.397 | HPRT | 0.188 |
| TFRC | 0.245 | RPL19 | 0.540 | GYPC | 0.190 |
| G6PD | 0.271 | HPRT | 0.702 | B2M | 0.212 |
| PGK1 | 0.282 | PGK1 | 0.915 | TFRC | 0.274 |
| GAPDH | 0.354 | TFRC | 0.941 | PGK1 | 0.280 |
| Gene name | Primers sequences (forward/reverse) | Spanned exons | Amplicon size (bp) | PCR efficiency (%) | R2 |
|---|---|---|---|---|---|
| ACTB | CCAACCGTGAGAAGATGACC | 2nd | 97 | 102.1 | 0.999 |
| CCAGAGGCGTACAGGGACAG | 3th | ||||
| GYPC | ATCAACATCGCTGTCATTGC | 3th | 117 | 106.7 | 0.994 |
| CTCGTTGGTGTGGTATGTGC | 4th | ||||
| RPL19 | AGCCTGTGACTGTCCATTCC | 2nd | 126 | 102.0 | 0.998 |
| ACGTTACCTTCTCGGGCATT | 3th | ||||
| GAPDH | CTGGCCAAGGTCATCCAT | 7th | 86 | 104.1 | 0.997 |
| ACAGTCTTCTGGGTGGCAGT | 8th | ||||
| YWHAZ | AGACGGAAGGTGCTGAGAAA | 2nd | 123 | 100.0 | 0.998 |
| CGTTGGGGATCAAGAACTTT | 3th | ||||
| PGK1 | ACTCCTTGCAGCCAGTTGCT | 3th | 101 | 109.9 | 0.991 |
| AGCACAAGCCTTCTCCACTTCT | 4th | ||||
| HPRT | TTTATTCCTCATGGACTAATTATGGA | 2nd | 71 | 99.8 | 0.987 |
| CCACCCATCTCCTTCATCAC | 3th | ||||
| TFRC | TTCTGGGCAGACCTCAAATC | 4th | 106 | 100.9 | 0.991 |
| CAGCTTCACGTGGGACATAA | 5th | ||||
| SDHA | CATCCACTACATGACGGAGCA | 4th | 90 | 99.8 | 0.996 |
| ATCTTGCCATCTTCAGTTCTGCTA | 5th | ||||
| G6PD | TGACCTATGGCAACCGATACAA | 10th | 76 | 103.0 | 0.990 |
| CCGCAAAAGACATCCAGGAT | 11th | ||||
| B2M | CTGTCGCTGTCTGGACTGG | 1st | 86 | 98.9 | 0.997 |
| TTTGGCTTTCCATCTTCTGG | 2nd |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Peletto, S.; Bertuzzi, S.; Campanella, C.; Modesto, P.; Maniaci, M.G.; Bellino, C.; Ariello, D.; Quasso, A.; Caramelli, M.; Acutis, P.L. Evaluation of Internal Reference Genes for Quantitative Expression Analysis by Real-Time PCR in Ovine Whole Blood. Int. J. Mol. Sci. 2011, 12, 7732-7747. https://doi.org/10.3390/ijms12117732
Peletto S, Bertuzzi S, Campanella C, Modesto P, Maniaci MG, Bellino C, Ariello D, Quasso A, Caramelli M, Acutis PL. Evaluation of Internal Reference Genes for Quantitative Expression Analysis by Real-Time PCR in Ovine Whole Blood. International Journal of Molecular Sciences. 2011; 12(11):7732-7747. https://doi.org/10.3390/ijms12117732
Chicago/Turabian StylePeletto, Simone, Simone Bertuzzi, Chiara Campanella, Paola Modesto, Maria Grazia Maniaci, Claudio Bellino, Dario Ariello, Antonio Quasso, Maria Caramelli, and Pier Luigi Acutis. 2011. "Evaluation of Internal Reference Genes for Quantitative Expression Analysis by Real-Time PCR in Ovine Whole Blood" International Journal of Molecular Sciences 12, no. 11: 7732-7747. https://doi.org/10.3390/ijms12117732