Effects of Different Force Fields and Temperatures on the Structural Character of Abeta (12–28) Peptide in Aqueous Solution


Abstract
:1. Introduction
2. Method
3. Results and Discussion
3.1. T-REMD Simulations
3.2. Distributions of Backbone Dihedral Angles of Aβ (12–28) Peptide
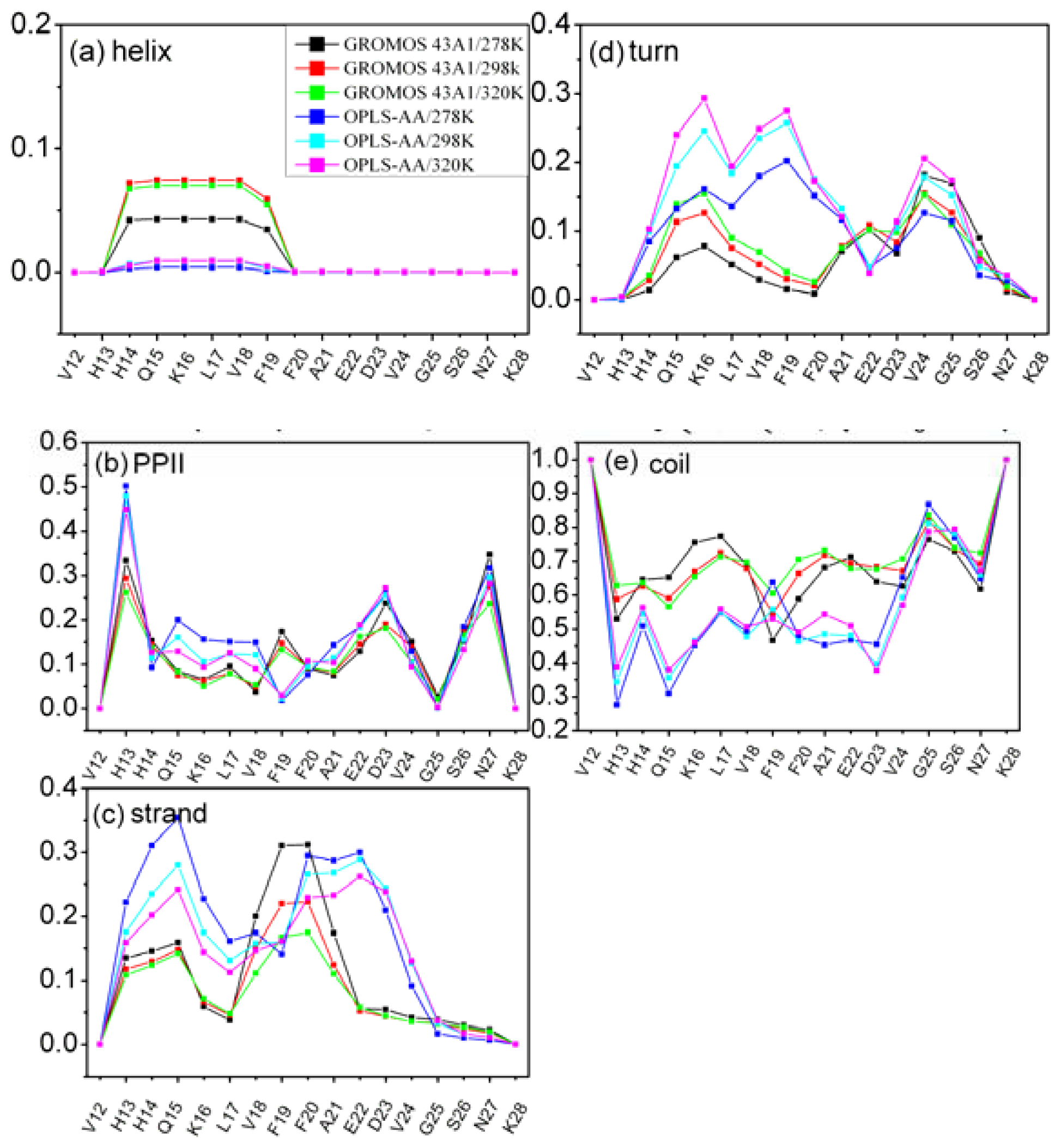
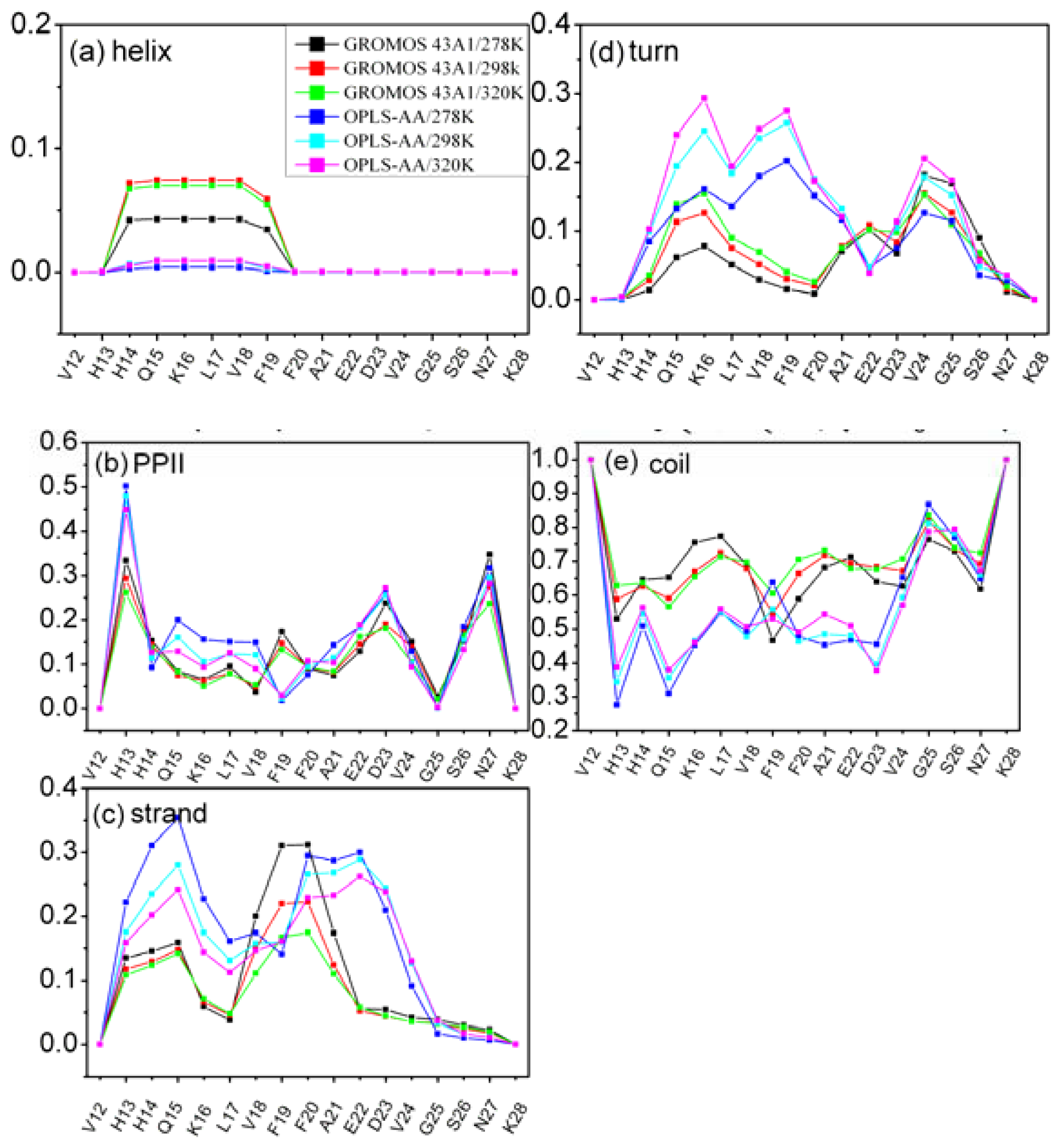
3.3. Residue Specific Secondary Structure Propensity of Aβ (12–28) Peptide
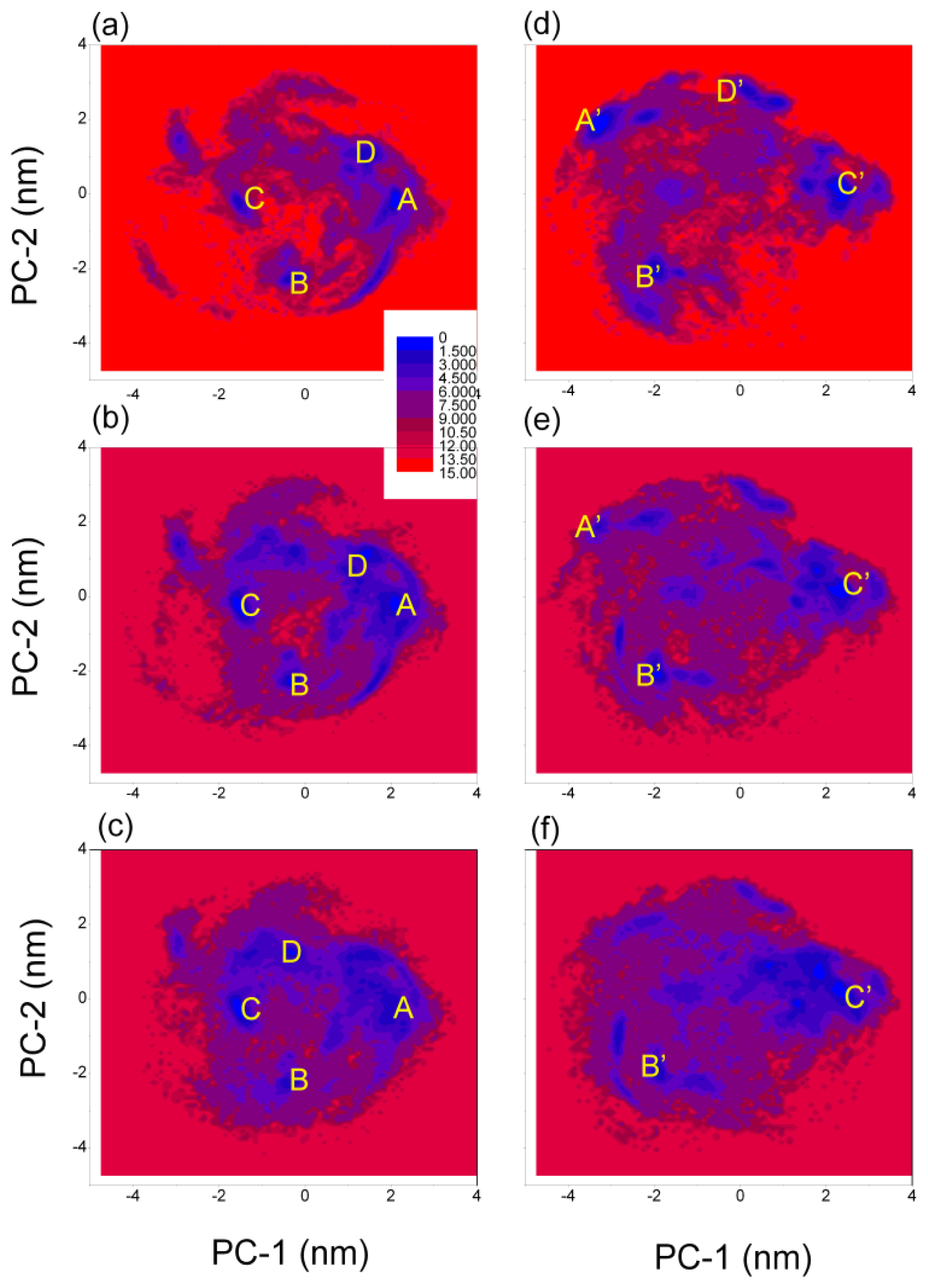
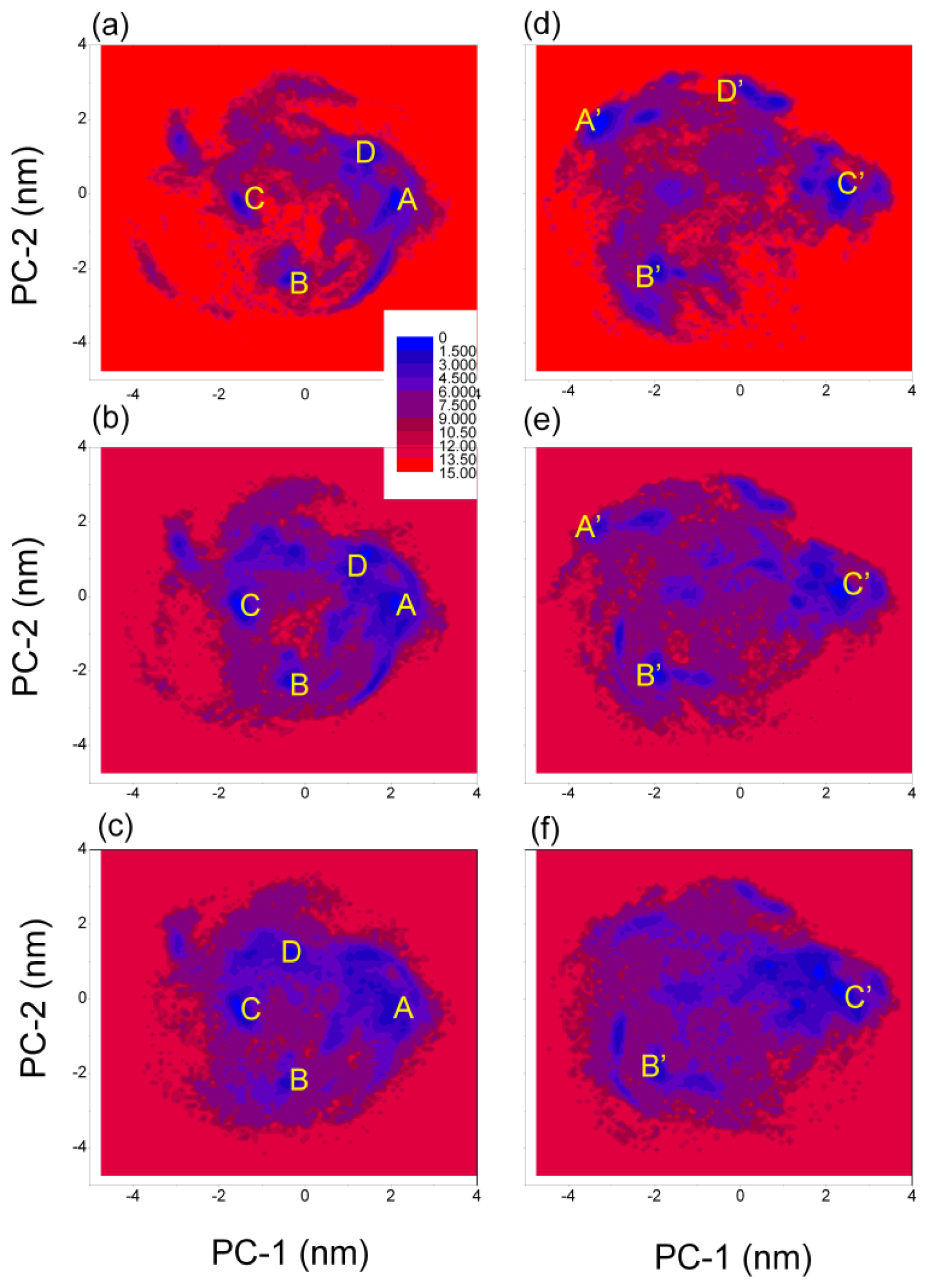
3.4. Free Energy Surface of Aβ (12–28) Peptide
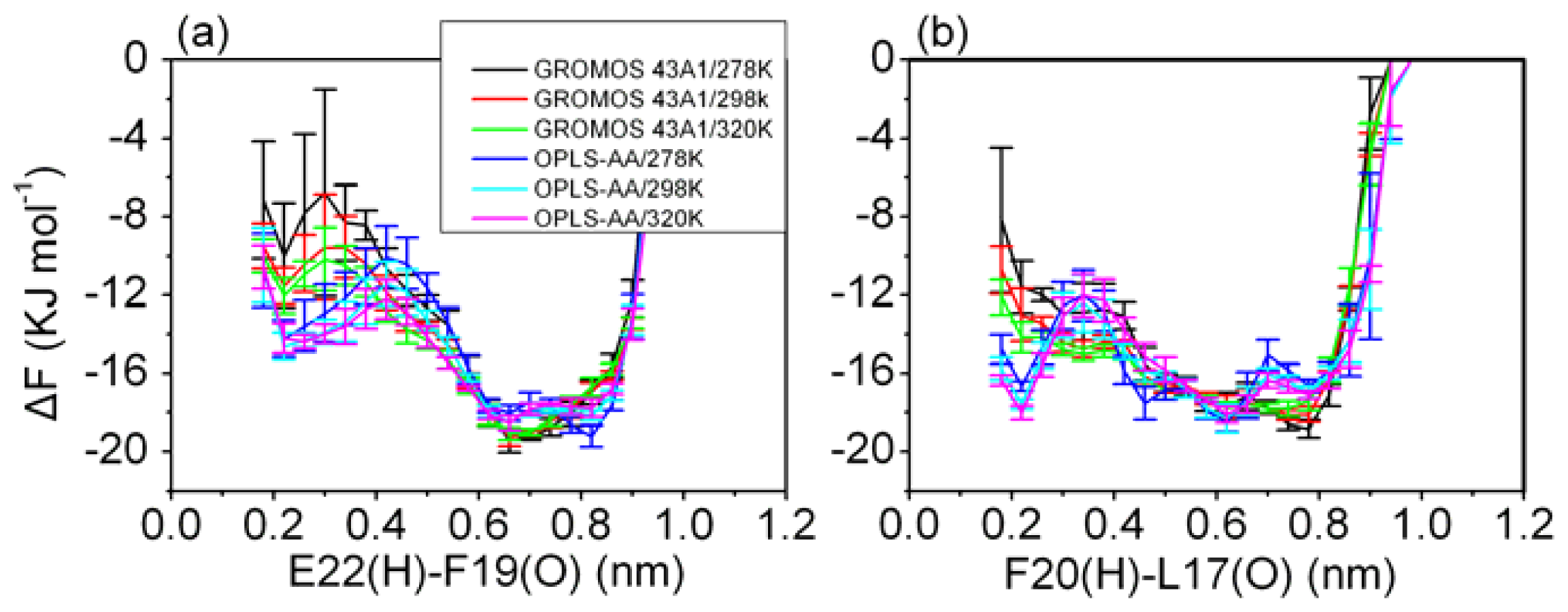
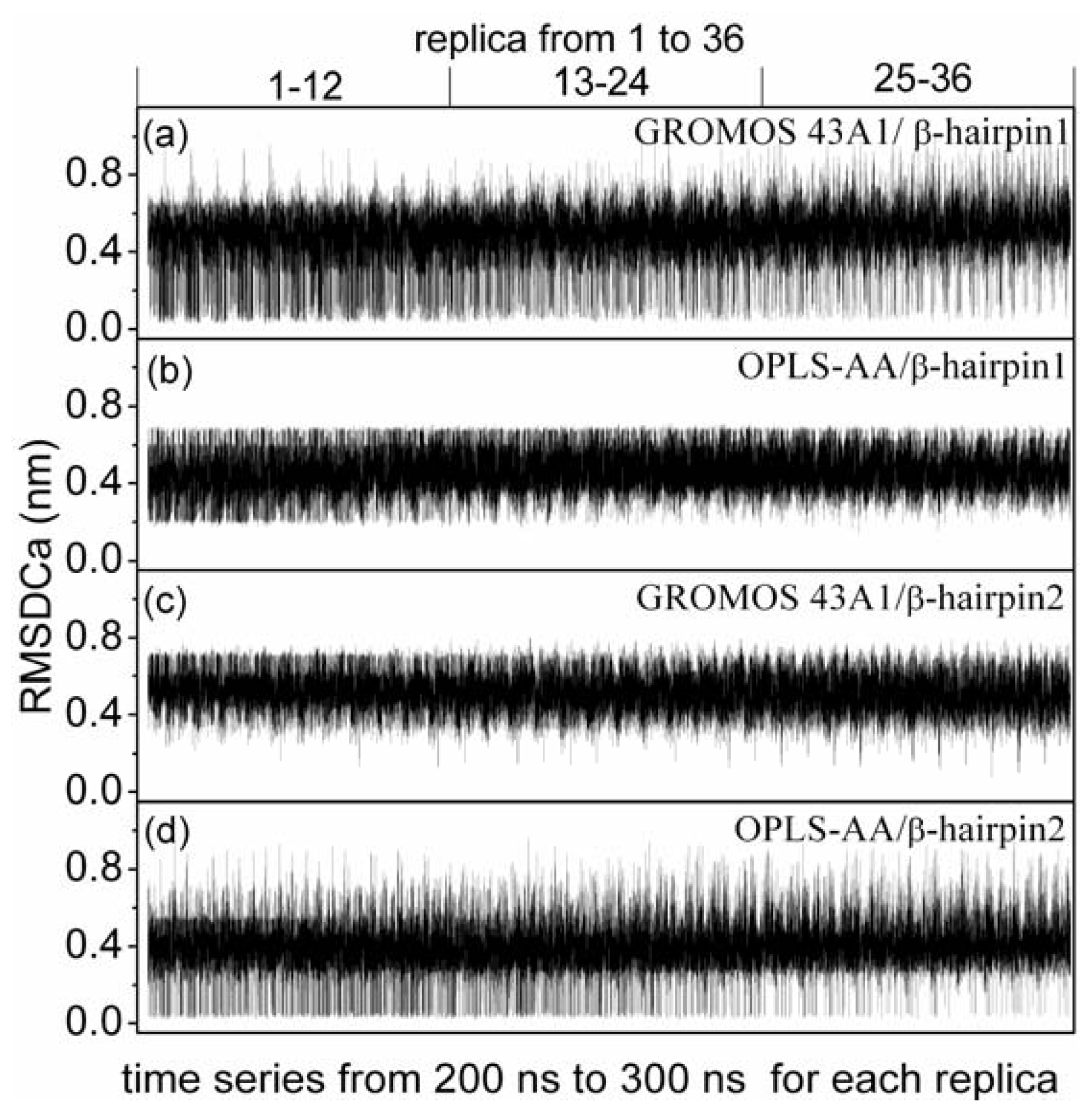
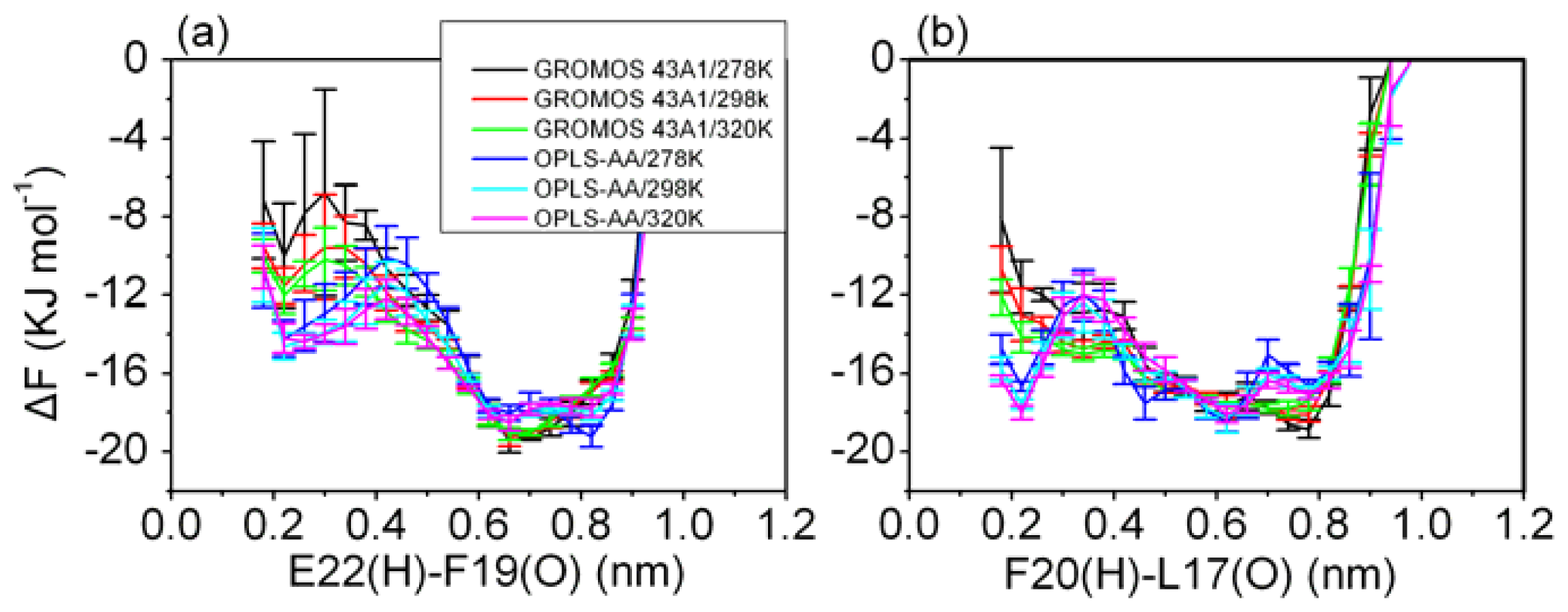
3.5. Formation of β-Hairpin in the Aβ (12–28) Peptide
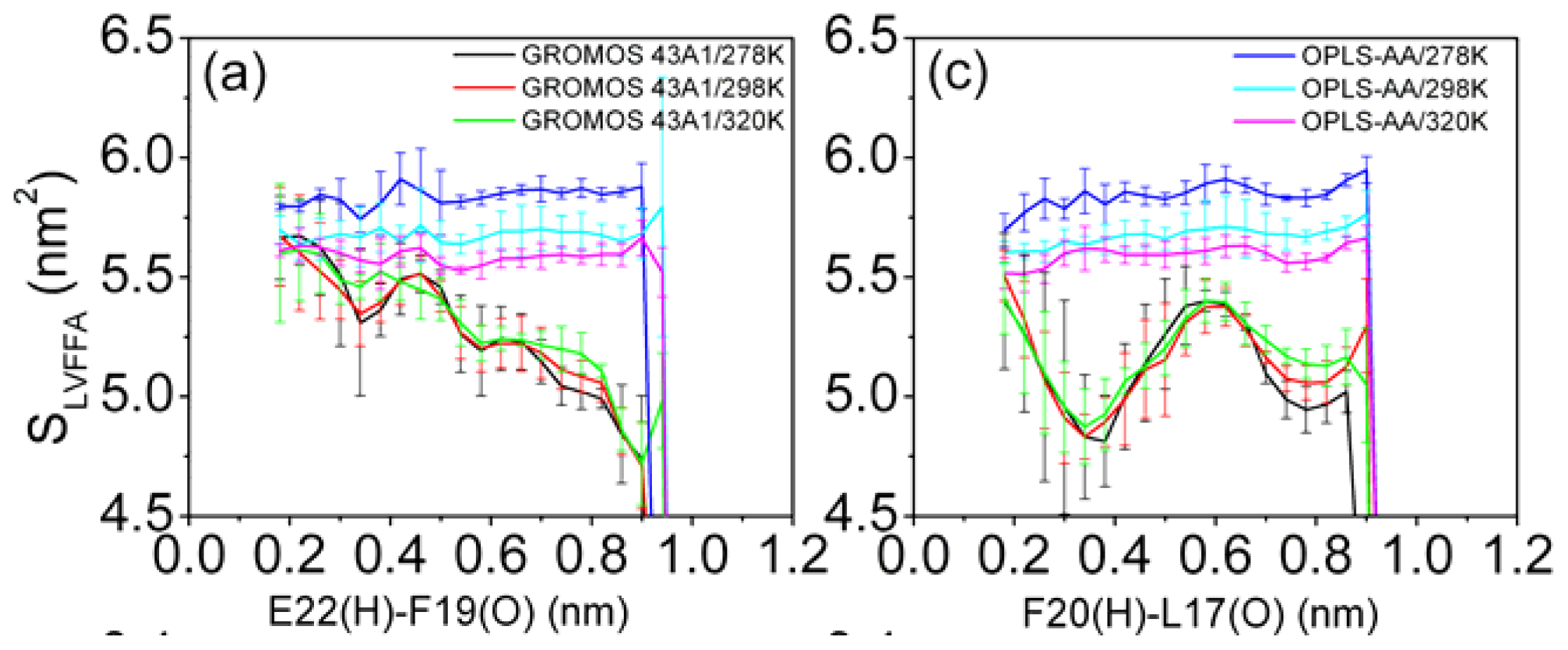
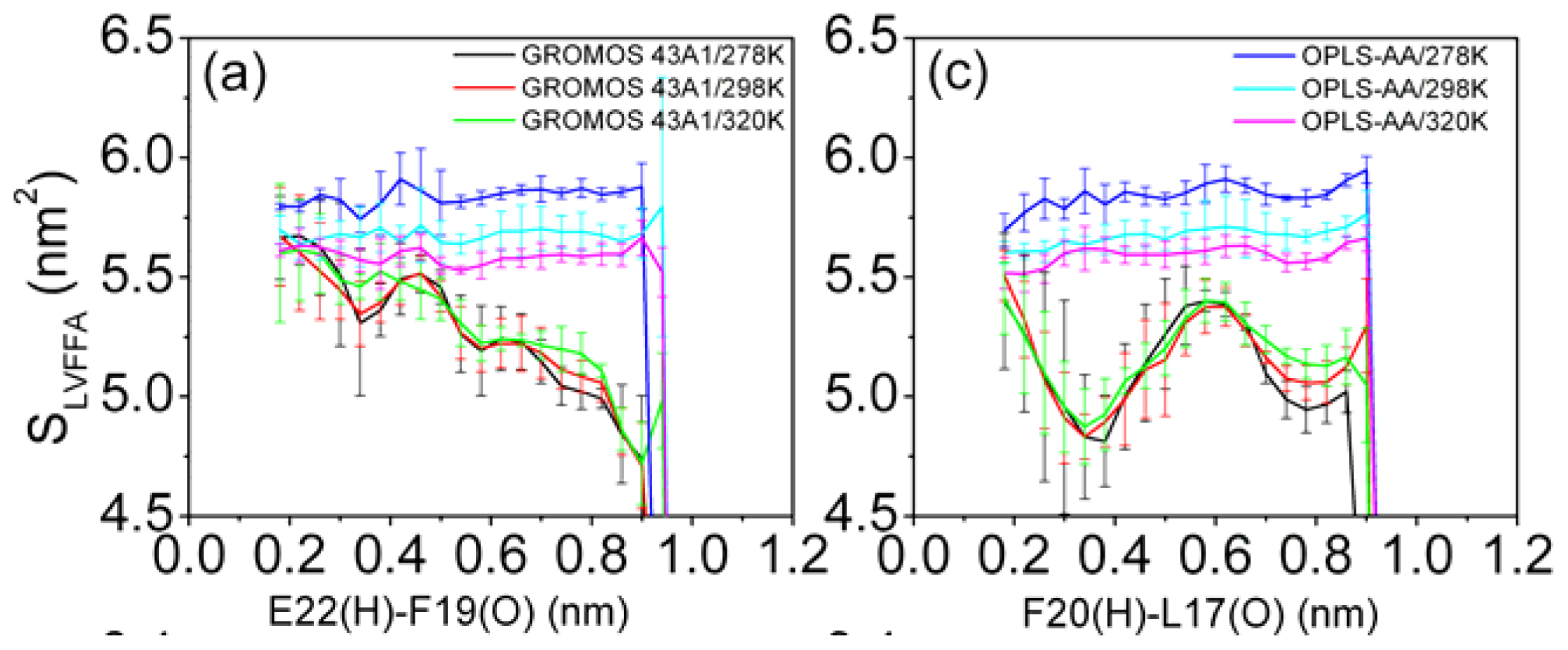
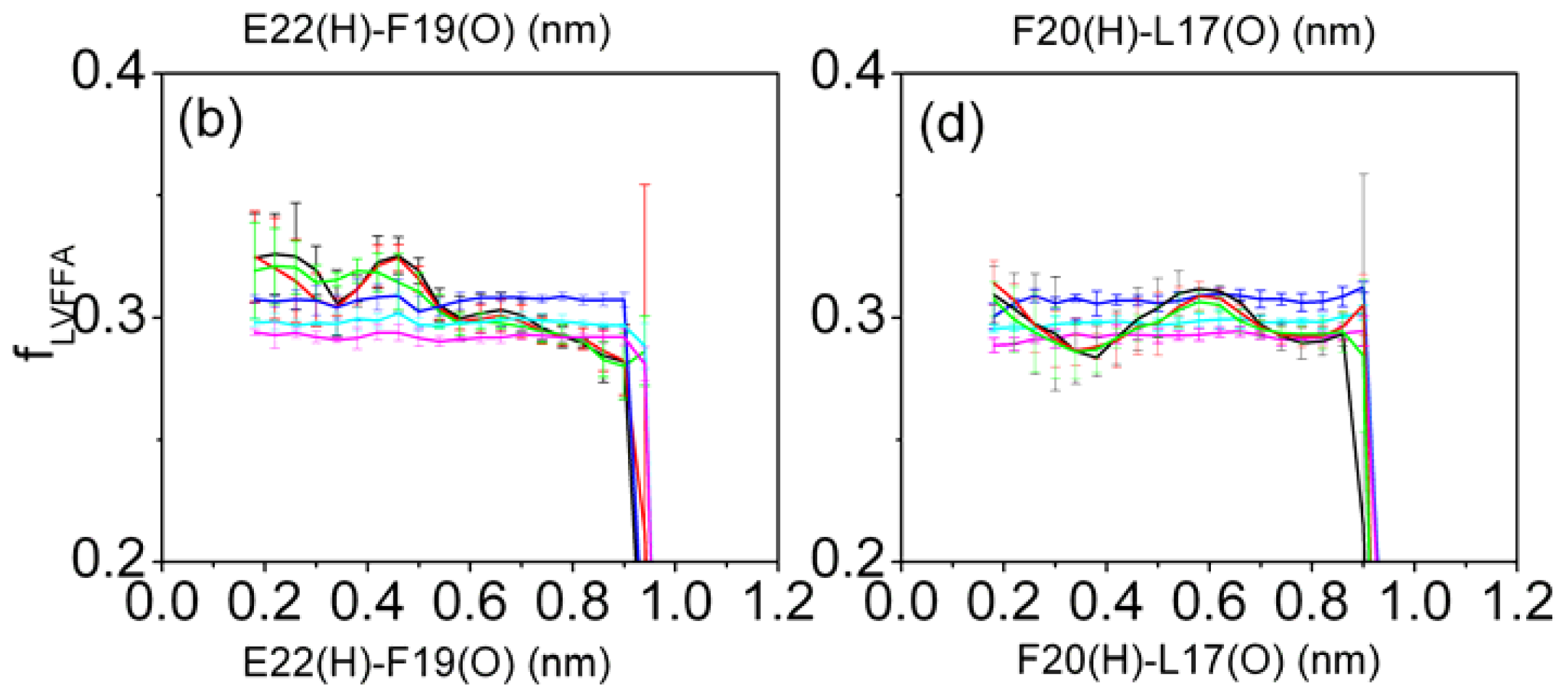
3.6. Solvent Exposure of Hydrophobic Residues in the Aβ (12–28) Peptide
4. Conclusions
Acknowledgments
References
- Pan, X.D.; Zhu, Y.G.; Lin, N.; Zhang, J.; Ye, Q.Y.; Huang, H.P.; Chen, X.C. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: Implications for Alzheimer’s disease. Mol. Neurodegener 2011, 6. [Google Scholar] [CrossRef]
- Sanchez, L.; Madurga, S.; Pukala, T.; Vilaseca, M.; Lopez-Iglesias, C.; Robinson, C.V.; Giralt, E.; Carulla, N. Abeta40 and Abeta42 amyloid fibrils exhibit distinct molecular recycling properties. J. Am. Chem. Soc 2011, 133, 6505–6508. [Google Scholar]
- Kumar, P.; Pillay, V.; Choonara, Y.E.; Modi, G.; Naidoo, D.; du Toit, L.C. In silico theoretical molecular modeling for Alzheimer’s disease: The nicotine-curcumin paradigm in neuroprotection and neurotherapy. Int. J. Mol. Sci 2011, 12, 694–724. [Google Scholar]
- Flood, J.F.; Morley, J.E.; Roberts, E. Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 3363–3366. [Google Scholar]
- Flood, J.F.; Morley, J.E.; Roberts, E. An amyloid beta-protein fragment, A beta [12–28], equipotently impairs post-training memory processing when injected into different limbic system structures. Brain Res 1994, 663, 271–276. [Google Scholar]
- Fraser, P.E.; Nguyen, J.T.; Surewicz, W.K.; Kirschner, D.A. pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys. J 1991, 60, 1190–1201. [Google Scholar]
- Fraser, P.E.; Levesque, L.; McLachlan, D.R. Alzheimer A beta amyloid forms an inhibitory neuronal substrate. J. Neurochem 1994, 62, 1227–1230. [Google Scholar]
- Fletcher, T.G.; Keire, D.A. The interaction of beta-amyloid protein fragment (12–28) with lipid environments. Protein Sci 1997, 6, 666–675. [Google Scholar]
- Danielsson, J.; Jarvet, J.; Damberg, P.; Graslund, A. The Alzheimer beta-peptide shows temperature-dependent transitions between left-handed 3-helix, beta-strand and random coil secondary structures. FEBS J 2005, 272, 3938–3949. [Google Scholar]
- Jarvet, J.; Damberg, P.; Danielsson, J.; Johansson, I.; Eriksson, L.E.; Graslund, A. A left-handed 3 (1) helical conformation in the Alzheimer Abeta (12–28) peptide. FEBS Lett 2003, 555, 371–374. [Google Scholar]
- Mackerell, A.D., Jr. Empirical force fields for biological macromolecules: Overview and issues. J. Comput. Chem. 2004, 25, 1584–1604. [Google Scholar]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glattli, A.; Hunenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. Engl 2006, 45, 4064–4092. [Google Scholar]
- Sorin, E.J.; Rhee, Y.M.; Shirts, M.R.; Pande, V.S. The solvation interface is a determining factor in peptide conformational preferences. J. Mol. Biol 2006, 356, 248–256. [Google Scholar]
- Hess, B.; van der Vegt, N.F. Hydration thermodynamic properties of amino acid analogues: A systematic comparison of biomolecular force fields and water models. J. Phys. Chem 2006, 110, 17616–17626. [Google Scholar]
- Reif, M.M.; Krautler, V.; Kastenholz, M.A.; Daura, X.; Hunenberger, P.H. Molecular dynamics simulations of a reversibly folding beta-heptapeptide in methanol: Influence of the treatment of long-range electrostatic interactions. J. Phys. Chem 2009, 113, 3112–3128. [Google Scholar]
- Nutt, D.R.; Smith, J.C. Molecular dynamics simulations of proteins: Can the explicit water model be varied? J. Chem. Theory Comput 2007, 3, 1550–1560. [Google Scholar]
- Day, R.; Paschek, D.; Garcia, A.E. Microsecond simulations of the folding/unfolding thermodynamics of the Trp-cage miniprotein. Proteins 2010, 78, 1889–1899. [Google Scholar]
- Gnanakaran, S.; Garcia, A.E. Helix-coil transition of alanine peptides in water: Force field dependence on the folded and unfolded structures. Proteins 2005, 59, 773–782. [Google Scholar]
- Matthes, D.; de Groot, B.L. Secondary structure propensities in peptide folding simulations: A systematic comparison of molecular mechanics interaction schemes. Biophysical. J 2009, 97, 599–608. [Google Scholar]
- Van Gunsteren, W.F.; Billeter, S.R.; Eising, A.A.; Hunenberger, P.H.; Krüger, P.; Mark, A.E.; Scott, W.R.P.; Tironi, I.G. Biomolecular Simulation: The GROMOS96 Manual and User Guide; vdf Hochschulverlag AG an der ETH Zürich and BIOMOS b.v.: Zürich, Switzerland and Groningen, The Netherlands, 1996. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS All-Atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- Wagoner, V.A.; Cheon, M.; Chang, I.; Hall, C.K. Computer simulation study of amyloid fibril formation by palindromic sequences in prion peptides. Proteins 2011, 79, 2132–2145. [Google Scholar]
- Natalello, A.; Benetti, F.; Doglia, S.M.; Legname, G.; Grandori, R. Compact conformations of alpha-synuclein induced by alcohols and copper. Proteins 2011, 79, 611–621. [Google Scholar]
- Nikolic, A.; Baud, S.; Rauscher, S.; Pomes, R. Molecular mechanism of beta-sheet self-organization at water-hydrophobic interfaces. Proteins 2011, 79, 1–22. [Google Scholar]
- Sgourakis, N.G.; Merced-Serrano, M.; Boutsidis, C.; Drineas, P.; Du, Z.; Wang, C.; Garcia, A.E. Atomic-level characterization of the ensemble of the Abeta (1–42) monomer in water using unbiased molecular dynamics simulations and spectral algorithms. J. Mol. Biol 2011, 405, 570–583. [Google Scholar]
- Daidone, I.; Simona, F.; Roccatano, D.; Broglia, R.A.; Tiana, G.; Colombo, G.; di Nola, A. Beta-hairpin conformation of fibrillogenic peptides: Structure and alpha-beta transition mechanism revealed by molecular dynamics simulations. Proteins 2004, 57, 198–204. [Google Scholar]
- Simona, F.; Tiana, G.; Broglia, R.A.; Colombo, G. Modeling the alpha-helix to beta-hairpin transition mechanism and the formation of oligomeric aggregates of the fibrillogenic peptide Abeta (12–28): Insights from all-atom molecular dynamics simulations. J. Mol. Graph. Model 2004, 23, 263–273. [Google Scholar]
- Baumketner, A.; Shea, J.E. Folding landscapes of the Alzheimer amyloid-beta (12–28) peptide. J. Mol. Biol 2006, 362, 567–579. [Google Scholar]
- Daidone, I.; Amadei, A.; di Nola, A. Thermodynamic and kinetic characterization of a beta-hairpin peptide in solution: An extended phase space sampling by molecular dynamics simulations in explicit water. Proteins 2005, 59, 510–518. [Google Scholar]
- Daidone, I.; Ulmschneider, M.B.; Di Nola, A.; Amadei, A.; Smith, J.C. Dehydration-driven solvent exposure of hydrophobic surfaces as a driving force in peptide folding. Proc. Natl. Acad. Sci. USA 2007, 104, 15230–15235. [Google Scholar]
- Munoz, V.; Ghirlando, R.; Blanco, F.J.; Jas, G.S.; Hofrichter, J.; Eaton, W.A. Folding and aggregation kinetics of a beta-hairpin. Biochemistry 2006, 45, 7023–7035. [Google Scholar]
- Cao, Z.; Wang, J. A comparative study of two different force fields on structural and thermodynamics character of H1 peptide via molecular dynamics simulations. J. Biomol. Struct. Dyn 2010, 27, 651–661. [Google Scholar]
- Cao, Z.; Wang, J. A comparative study of different temperatures on computed structural character of H1 peptide via temperature replica exchange molecular dynamics simulations. Prog. Biochem. Biophys 2010, 37, 319–325. [Google Scholar]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. Intermol. Forces 1981, 331–342. [Google Scholar]
- Cao, Z.; Liu, L.; Wu, P.; Wang, J. Structural and thermodynamics characters of isolated α-syn12 peptide: Long-time temperature replica-exchange molecular dynamics in aqueous solution. Acta Biochim. Biophys. Sin 2011, 43, 172–180. [Google Scholar]
- Cao, Z.; Liu, L.; Wang, J. Effects of pH and temperature on the structural and thermodynamic character of alpha-syn12 peptide in aqueous solution. J. Biomol. Struct. Dyn 2010, 28, 343–353. [Google Scholar]
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impy, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput 2008, 4, 435–447. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N. log(N) method for Ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys 1977, 23((77)), 90098–5. [Google Scholar] [CrossRef]
- Patriksson, A.; van der Spoel, D. A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys 2008, 10, 2073–2077. [Google Scholar]
- Hu, H.; Elstner, M.; Hermans, J. Comparison of a QM/MM force field and molecular mechanics force fields in simulations of alanine and glycine “dipeptides” (Ace-Ala-Nme and Ace-Gly-Nme) in water in relation to the problem of modeling the unfolded peptide backbone in solution. Proteins 2003, 50, 451–463. [Google Scholar]
- Srinivasan, R.; Rose, G.D. A physical basis for protein secondary structure. Proc. Natl. Acad. Sci. USA 1999, 96, 14258–14263. [Google Scholar]
- Jarvet, J.; Damberg, P.; Bodell, K.; Eriksson, L.E.G.; Gräslund, A. Reversible random coil to β-sheet transition and the early stage of aggregation of the Aβ (12–28) fragment from the alzheimer peptide. J. Am. Chem. Soc 2000, 122, 4261–4268. [Google Scholar]
- Garcia, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett 1992, 68, 2696–2699. [Google Scholar]
- Nguyen, J.; Baldwin, M.A.; Cohen, F.E.; Prusiner, S.B. Prion protein peptides induce alpha-helix to beta-sheet conformational transitions. Biochemistry 1995, 34, 4186–4192. [Google Scholar]
- Inouye, H.; Kirschner, D.A. Polypeptide chain folding in the hydrophobic core of hamster scrapie prion: Analysis by X-ray diffraction. J. Struct. Biol 1998, 122, 247–255. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GROMOS 43A1/SPC T = 278 K | GROMOS 43A1/SPC T = 298 K | GROMOS 43A1/SPC T = 320 K | OPLS-AA/TIP4P T = 278 K | OPLS-AA/TIP4P T = 298 K | OPLS-AA/TIP4P T = 320 K | |
|---|---|---|---|---|---|---|
| α region | 0.26 (0.26) | 0.28 (0.27) | 0.30 (0.29) | 0.30 | 0.33 | 0.34 |
| Bridge region | 0.06 (0.07) | 0.07 (0.08) | 0.08 (0.08) | 0.06 | 0.07 | 0.07 |
| β region | 0.62 (0.61) | 0.58 (0.59) | 0.55 (0.57) | 0.62 | 0.58 | 0.56 |
| GROMOS 43A1/SPC T = 278 K | GROMOS 43A1/SPC T = 298 K | GROMOS 43A1/SPC T = 320 K | OPLS-AA/TIP4P T = 278 K | OPLS-AA/TIP4P T = 298 K | OPLS-AA/TIP4P T = 320 K | |
|---|---|---|---|---|---|---|
| α-helix | 0.01 | 0.02 | 0.02 | 0.00 | 0.00 | 0.00 |
| PPII | 0.13 | 0.12 | 0.11 | 0.15 | 0.14 | 0.13 |
| β-strand | 0.10 | 0.08 | 0.07 | 0.16 | 0.15 | 0.14 |
| β-turn | 0.06 | 0.06 | 0.07 | 0.09 | 0.12 | 0.13 |
| coil | 0.70 | 0.71 | 0.72 | 0.59 | 0.59 | 0.60 |
| (PC-1, PC-2) coordinates (nm) and relative depths (kJ mol−1) of the minima | ||||
|---|---|---|---|---|
| A | B | C | D | |
| GROMOS 43A1/SPC | (2.05, −0.25) | (−0.35, −2.25) | (−1.55, −0.15) | (1.35, 0.95) |
| T = 278 K | 0.0 | 1.4 | 2.0 | 1.8 |
| GROMOS 43A1/SPC | (2.15, −0.15) | (−0.35, −2.25) | (−1.45, −0.35) | (1.45, 1.15) |
| T = 298 K | 0.5 | 0.4 | 0.0 | 0.9 |
| GROMOS 43A1/SPC | (2.15, −0.25) | (−0.45, −2.25) | (−1.55, −0.25) | (−0.35, 1.25) |
| T = 320 K | 1.5 | 1.5 | 0.0 | 0.9 |
| A′ | B′ | C′ | D′ | |
| OPLS-AA/TIP4P | (−3.25, 1.95) | (−1.95, −2.15) | (2.35, 0.25) | (0.25, 2.75) |
| T = 278 K | 0.0 | 1.1 | 1.0 | 1.6 |
| OPLS-AA/TIP4P | (−3.35, 1.85) | (−1.95, −2.05) | (2.35, 0.25) | |
| T = 298 K | 2.3 | 0.7 | 0.0 | |
| OPLS-AA/TIP4P | (−1.95, −1.95) | (2.45, 0.15) | ||
| T = 320 K | 1.4 | 0.0 | ||
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cao, Z.; Liu, L.; Zhao, L.; Wang, J. Effects of Different Force Fields and Temperatures on the Structural Character of Abeta (12–28) Peptide in Aqueous Solution. Int. J. Mol. Sci. 2011, 12, 8259-8274. https://doi.org/10.3390/ijms12118259
Cao Z, Liu L, Zhao L, Wang J. Effects of Different Force Fields and Temperatures on the Structural Character of Abeta (12–28) Peptide in Aqueous Solution. International Journal of Molecular Sciences. 2011; 12(11):8259-8274. https://doi.org/10.3390/ijms12118259
Chicago/Turabian StyleCao, Zanxia, Lei Liu, Liling Zhao, and Jihua Wang. 2011. "Effects of Different Force Fields and Temperatures on the Structural Character of Abeta (12–28) Peptide in Aqueous Solution" International Journal of Molecular Sciences 12, no. 11: 8259-8274. https://doi.org/10.3390/ijms12118259