Coenzyme Q10 Ameliorates Ultraviolet B Irradiation Induced Cell Death Through Inhibition of Mitochondrial Intrinsic Cell Death Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
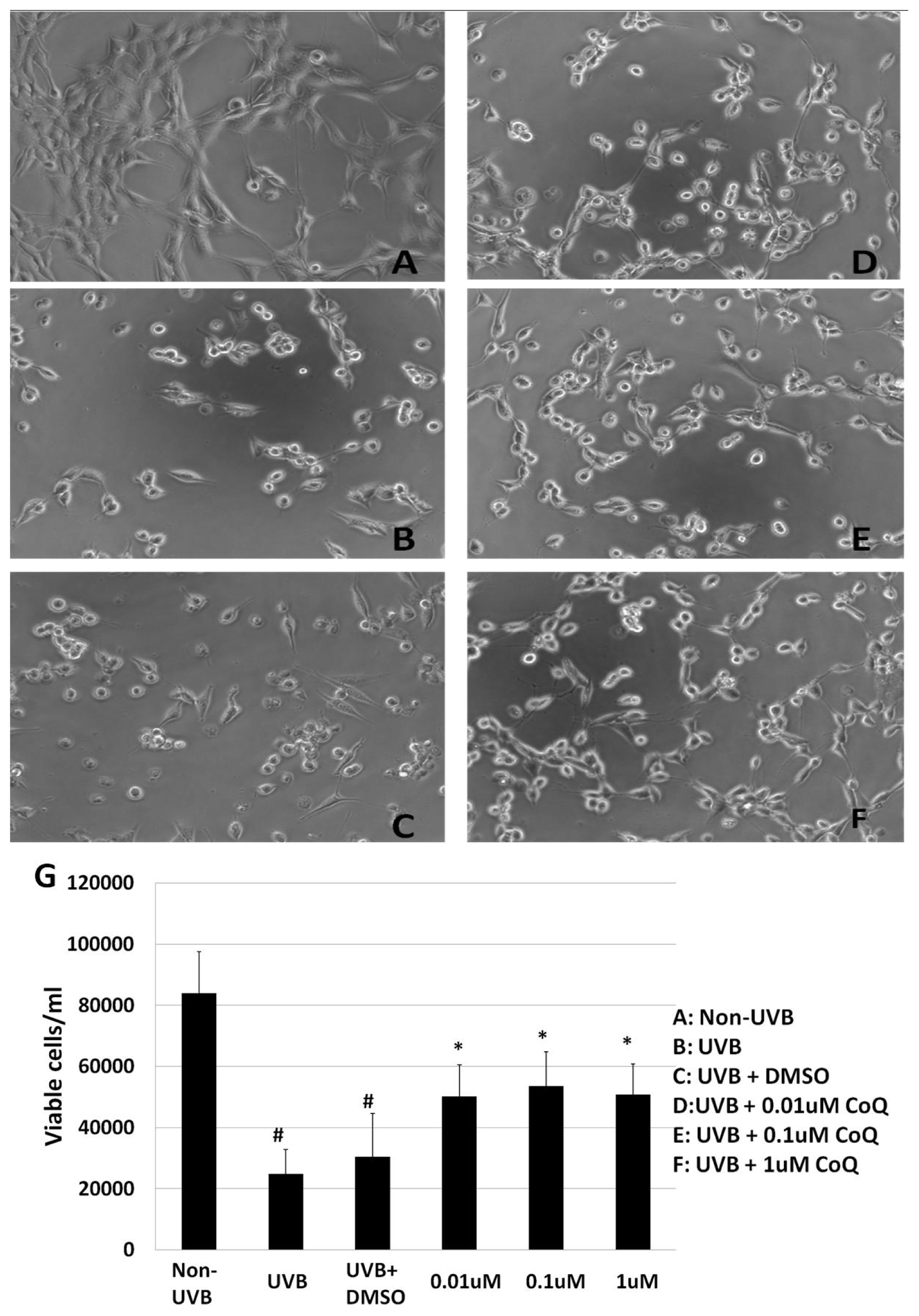
2.1. Effect of CoQ10 on UVB Irradiation Induced HT22 Cells Death
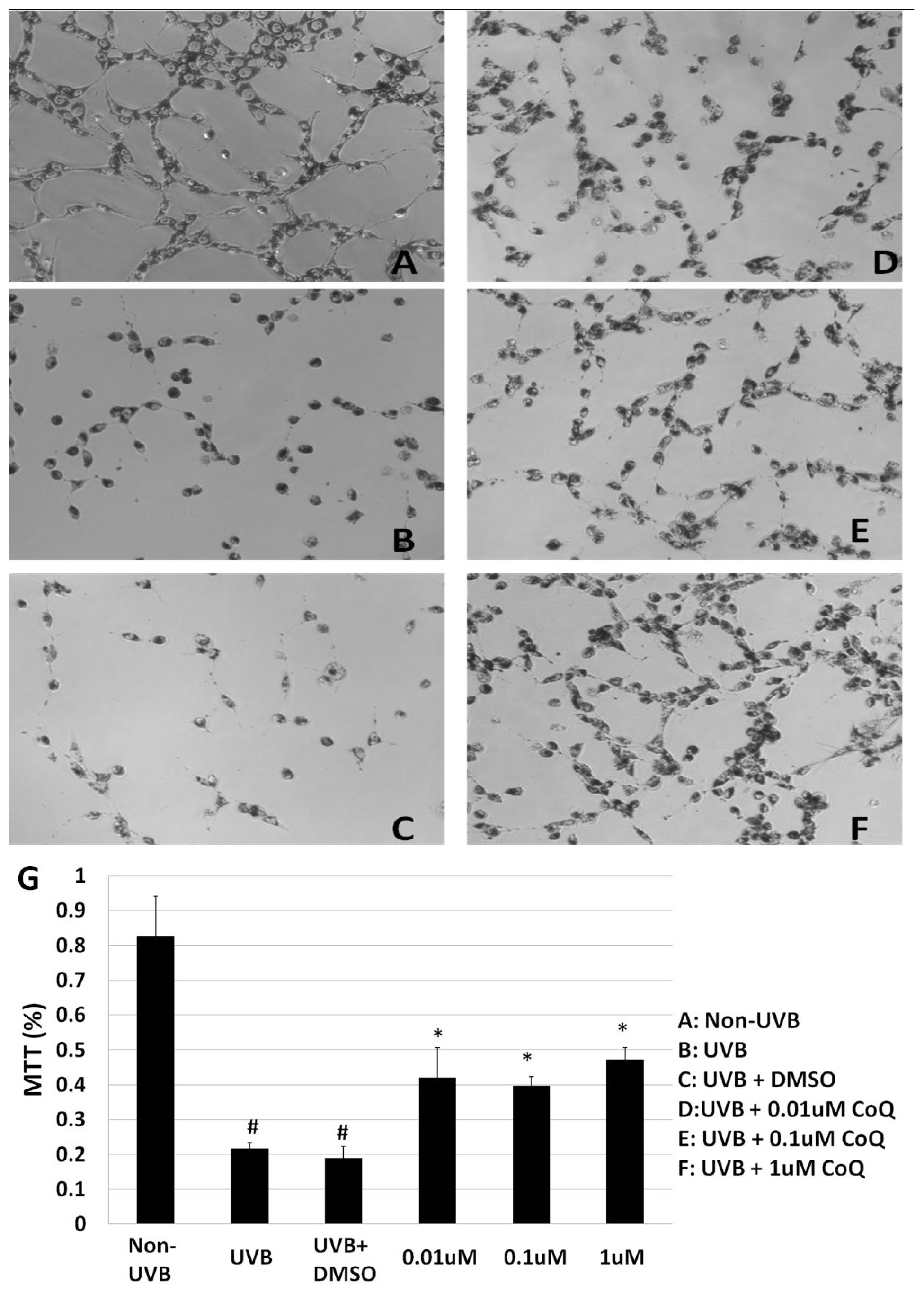
2.2. Influence of CoQ10 on Mitochondrial Succinate Dehydrogenase Activity
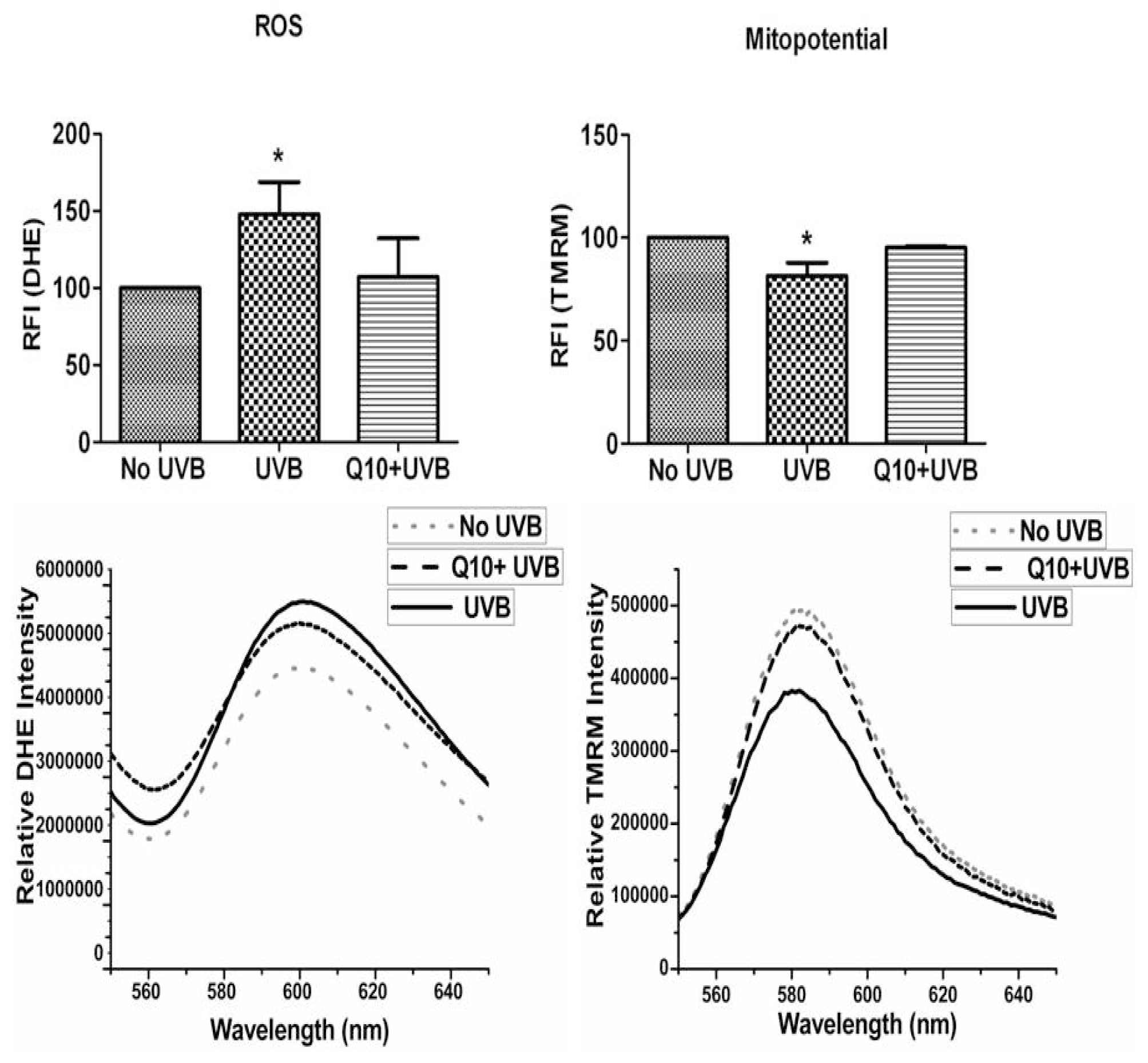
2.3. Effects of CoQ10 on ROS Production and Mitochondrial Membrane Potential
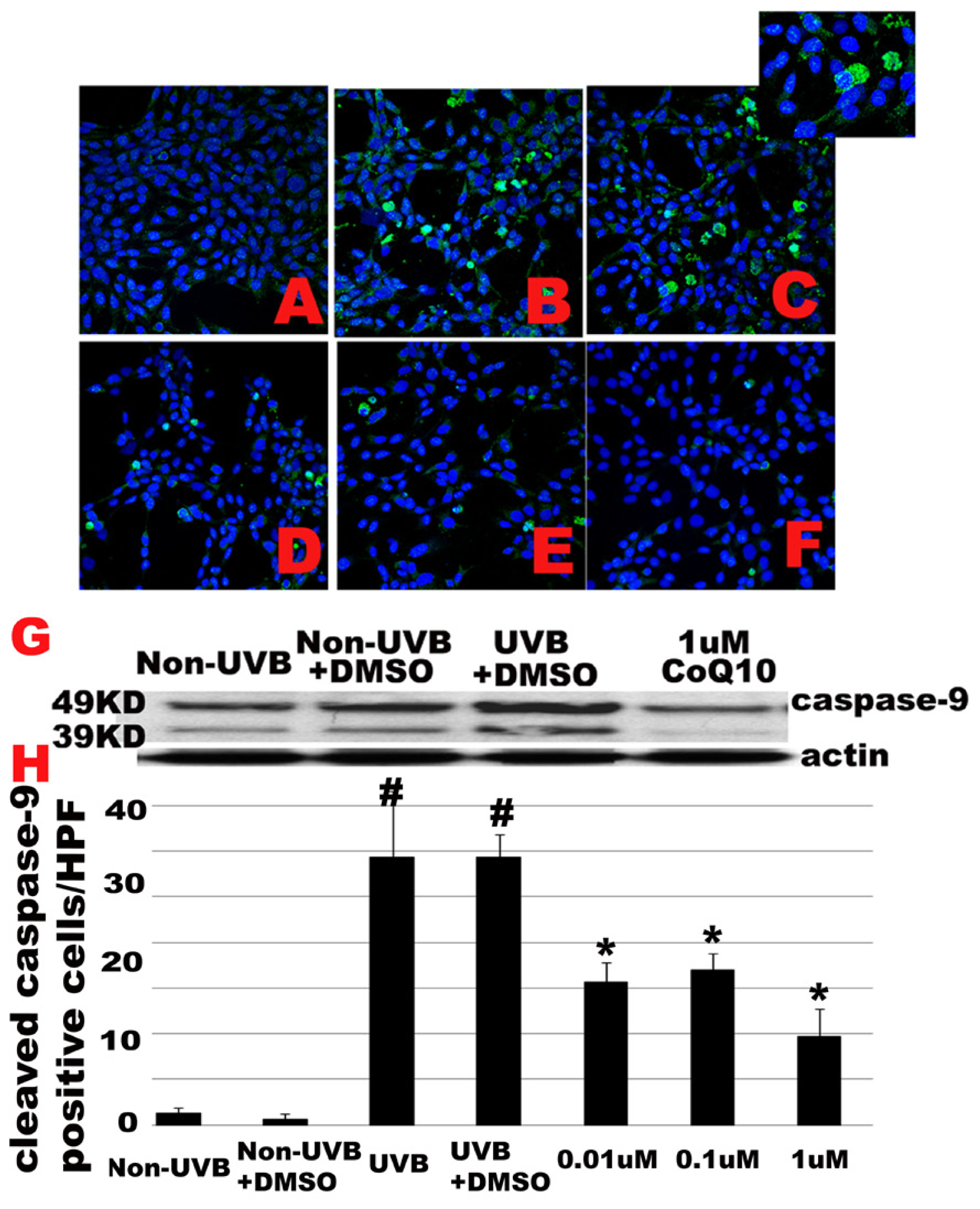
2.4. Effect of CoQ10 on UVB Irradiation Induced Caspase-9 Activation
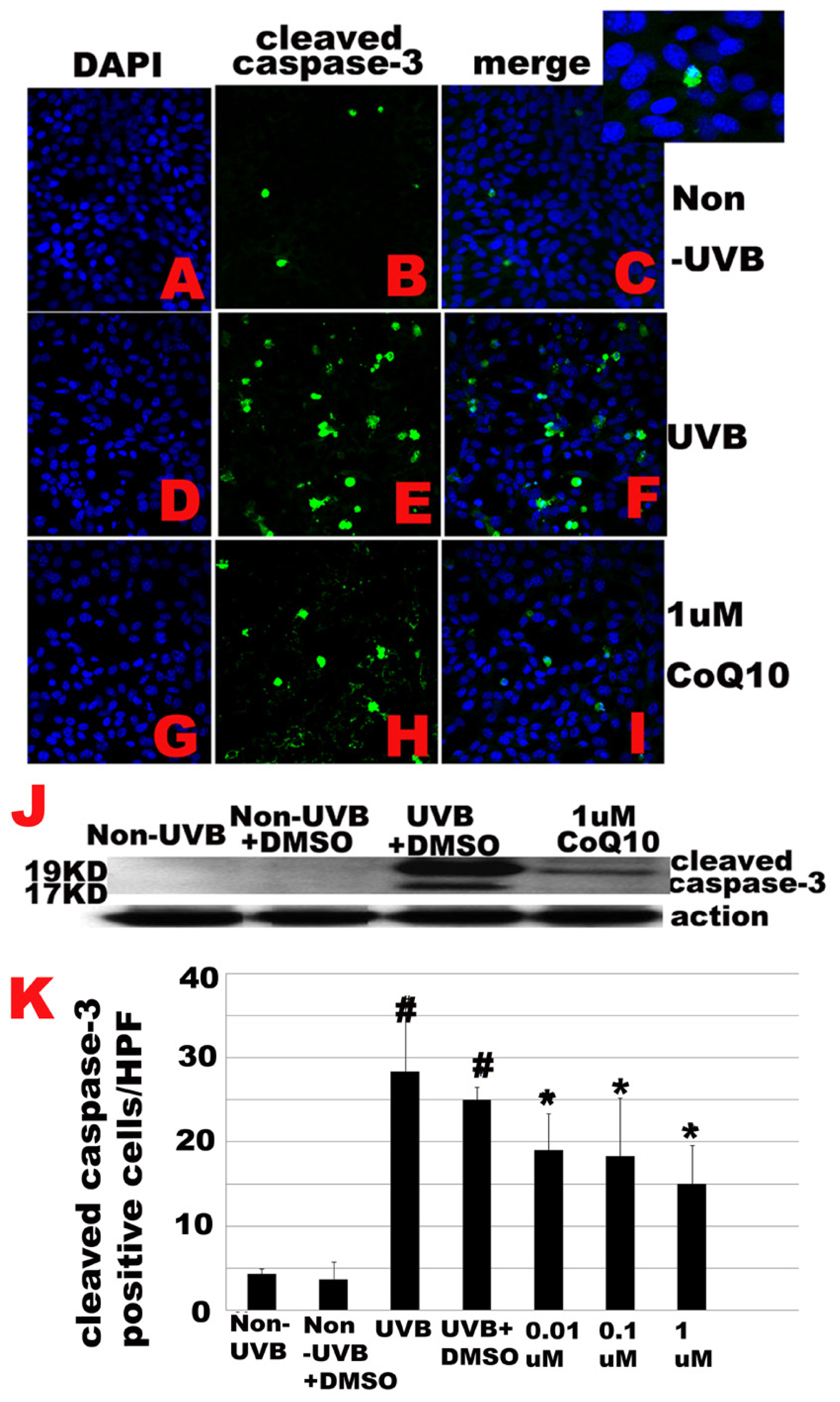
2.5. Effect of CoQ10 on UVB Irradiation Induced Caspase-3 Activation
2.6. Discussion
3. Material and Methods
3.1. Materials
3.2. Cell Culture, UVB Irradiation and CoQ10 Treatment
3.3. Cell Viability Assay
3.4. Mitochondrial Succinate Dehydrogenase Activity
3.5. Measurements of Superoxide and Mitochondrial Membrane Potential
3.6. Western Blot Analysis
3.7. Immunocytochemistry
3.8. Statistical Analysis
4. Conclusion
Acknowledgements
References
- Trabosh, V.A.; Daher, A.; Divito, K.A.; Amin, K.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S. UVB upregulates the bax promoter in immortalized human keratinocytes via ROS induction of Id3. Exp. Dermatol 2009, 18, 387–395. [Google Scholar]
- Park, L.J.; Ju, S.M.; Song, H.Y.; Lee, J.A.; Yang, M.Y.; Kang, Y.H.; Kwon, H.J.; Kim, T.Y.; Choi, S.Y.; Park, J. The enhanced monocyte adhesiveness after UVB exposure requires ROS and NF-kappaB signaling in human keratinocyte. J. Biochem. Mol. Biol 2006, 39, 618–625. [Google Scholar]
- Benitez-Alfonso, Y.; Jackson, D.; Maule, A. Redox regulation of intercellular transport. Protoplasma 2011, 248, 131–140. [Google Scholar]
- Kim, C.H.; Jeon, H.M.; Lee, S.Y.; Jeong, E.K.; Ju, M.K.; Park, B.J.; Park, H.G.; Lim, S.C.; Han, S.I.; Kang, H.S. Role of reactive oxygen species-dependent protein aggregation in metabolic stress-induced necrosis. Int. J. Oncol 2010, 37, 97–102. [Google Scholar]
- Myatt, S.S.; Brosens, J.J.; Lam, E.W. Sense and sensitivity: FOXO and ROS in cancer development and treatment. Antioxid. Redox Signal 2011, 14, 675–687. [Google Scholar]
- Mendelev, N.; Witherspoon, S.; Li, P.A. Overexpression of human selenoprotein H in neuronal cells ameliorates ultraviolet irradiation-induced damage by modulating cell signaling pathways. Exp. Neurol 2009, 220, 328–334. [Google Scholar]
- Mendelev, N.; Mehta, S.L.; Witherspoon, S.; He, Q.; Sexton, J.Z.; Li, P.A. Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion 2011, 11, 76–82. [Google Scholar]
- Hargreaves, I.P.; Heales, S.J.; Briddon, A.; Land, J.M.; Lee, P.J. Blood mononuclear cell coenzyme Q10 concentration and mitochondrial respiratory chain succinate cytochrome-c reductase activity in phenylketonuric patients. J. Inherit. Metab. Dis 2002, 25, 673–679. [Google Scholar]
- Sohal, R.S.; Forster, M.J. Coenzyme Q, oxidative stress and aging. Mitochondrion 2007, 7, 103–111. [Google Scholar]
- Kumar, A.; Kaur, H.; Devi, P.; Mohan, V. Role of coenzyme Q10 (CoQ10) in cardiac disease, hypertension and Meniere-like syndrome. Pharmacol. Ther 2009, 124, 259–268. [Google Scholar]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spanò, A.; Cavaliere, F.; Rombolà, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal damage caused by high intraocular pressure-induced transient ischemia is prevented by coenzyme Q10 in rat. Int. Rev. Neurobiol 2007, 82, 397–406. [Google Scholar]
- Hoppe, U.; Bergemann, J.; Diembeck, W.; Ennen, J.; Gohla, S.; Harris, I.; Jacob, J.; Kielholz, J.; Mei, W.; Pollet, D.; et al. Coenzyme Q10, a cutaneous antioxidant and energizer. Biofactors 1999, 9, 371–378. [Google Scholar]
- Ben Jilani, K.E.; Panee, J.; He, Q.; Berry, M.J.; Li, P.A. Overexpression of selenoprotein H reduces Ht22 neuronal cell death after UVB irradiation by preventing superoxide formation. Int. J. Biol. Sci 2007, 3, 198–204. [Google Scholar]
- Littarru, G.P.; Tiano, L. Bioenergetic and antioxidant properties of coenzyme Q10: Recent developments. Mol. Biotechnol 2007, 37, 31–37. [Google Scholar]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem 2003, 278, 28220–28228. [Google Scholar]
- Prahl, S.; Keuper, T.; Biernoth, T.; Wohrmann, Y.; Munster, A.; Furstenau, M; Schmidt, M.; Schulze, C.; Witter, K.-P.; Wenk, H.; et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. Biofactors 2008, 32, 245–255. [Google Scholar]
- Muta-Takada, K.; Terada, T.; Yamanishi, H.; Ashida, Y.; Inomata, S.; Nishiyama, T.; Amano, S. Coenzyme Q10 protects against oxidative stress-induced cell death and enhances the synthesis of basement membrane components in dermal and epidermal cells. Biofactors 2009, 35, 435–441. [Google Scholar]
- Inui, M.; Ooe, M.; Fujii, K.; Matsunaka, H.; Yoshida, M.; Ischihashi, M. Mechnisms of inhibitory effects of CoQ10 on UVB-induced wrinkle formation in vitro and in vivo. Biofactors 2008, 32, 237–243. [Google Scholar]
- Somayajulu, M.; McCarthy, S.; Hung, M.; Sikorska, M.; Borowy-Borowski, H.; Pandey, S. Role of mitochondria in neuronal cell death induced by oxidative stress; neuroprotection by Coenzyme Q10. Neurobiol. Dis 2005, 18, 618–627. [Google Scholar]
- Somayajulu-Niţu, M.; Sandhu, J.K.; Cohen, J.; Sikorska, M.; Sridhar, T.S.; Matei, A.; Borowy-Borowski, H.; Pandey, S. Paraquat induces oxidative stress, neuronal loss in substantia nigra region and parkinsonism in adult rats: Neuroprotection and amelioration of symptoms by water-soluble formulation of coenzyme Q10. BMC Neurosci 2009, 10, 88. [Google Scholar]
- Abdin, A.A.; Hamouda, H.E. Mechanism of the neuroprotective role of coenzyme Q10 with or without L-dopa in rotenone-induced parkinsonism. Neuropharmacology 2008, 55, 1340–1346. [Google Scholar]
- Li, G.; Jack, C.R.; Yang, X.F.; Yang, E.S. Diet supplement CoQ10 delays brain atrophy in aged transgenic mice with mutations in the amyloid precursor protein: An in vivo volume MRI study. Biofactors 2008, 32, 169–178. [Google Scholar]
- Naderi, J.; Somayajulu-Nitu, M.; Mukerji, A.; Sharda, P.; Sikorska, M.; Borowy-Borowski, H.; Antonsson, B.; Pandey, S. Water-soluble formulation of Coenzyme Q10 inhibits Bax-induced destabilization of mitochondria in mammalian cells. Apoptosis 2006, 11, 1359–1369. [Google Scholar]
- Schmelzer, C.; Lindner, I.; Rimbach, G.; Niklowitz, P.; Menke, T.; Döring, F. Functions of coenzyme Q10 in inflammation and gene expression. Biofactors 2008, 32, 179–183. [Google Scholar]





© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jing, L.; Kumari, S.; Mendelev, N.; Li, P.A. Coenzyme Q10 Ameliorates Ultraviolet B Irradiation Induced Cell Death Through Inhibition of Mitochondrial Intrinsic Cell Death Pathway. Int. J. Mol. Sci. 2011, 12, 8302-8315. https://doi.org/10.3390/ijms12118302
Jing L, Kumari S, Mendelev N, Li PA. Coenzyme Q10 Ameliorates Ultraviolet B Irradiation Induced Cell Death Through Inhibition of Mitochondrial Intrinsic Cell Death Pathway. International Journal of Molecular Sciences. 2011; 12(11):8302-8315. https://doi.org/10.3390/ijms12118302
Chicago/Turabian StyleJing, Li, Santosh Kumari, Natalia Mendelev, and P. Andy Li. 2011. "Coenzyme Q10 Ameliorates Ultraviolet B Irradiation Induced Cell Death Through Inhibition of Mitochondrial Intrinsic Cell Death Pathway" International Journal of Molecular Sciences 12, no. 11: 8302-8315. https://doi.org/10.3390/ijms12118302