Predicting Antitumor Activity of Peptides by Consensus of Regression Models Trained on a Small Data Sample

Abstract
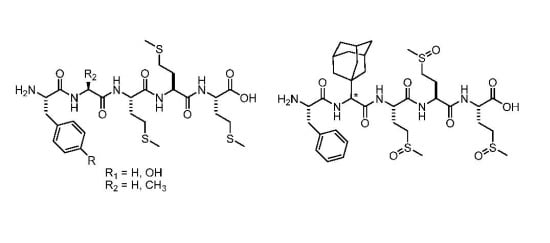
:
1. Introduction
2. Results and Discussion
2.1. Regression Model Selection for Compound Activity Prediction
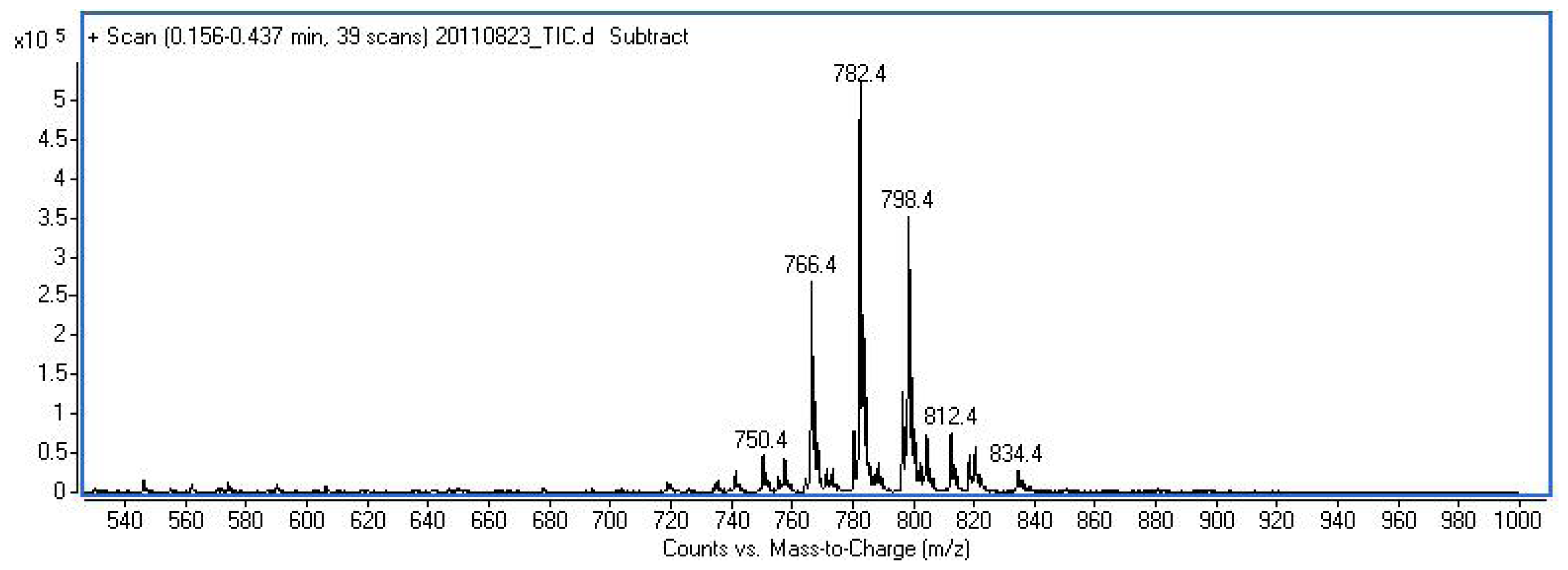
2.2. Peptide Synthesis
2.3. Proliferation Assay
3. Experimental Section
3.1. Construction of the Virtual Library
3.2. Software Environment
3.3. Peptide Synthesis
3.3.1. Materials and Methods
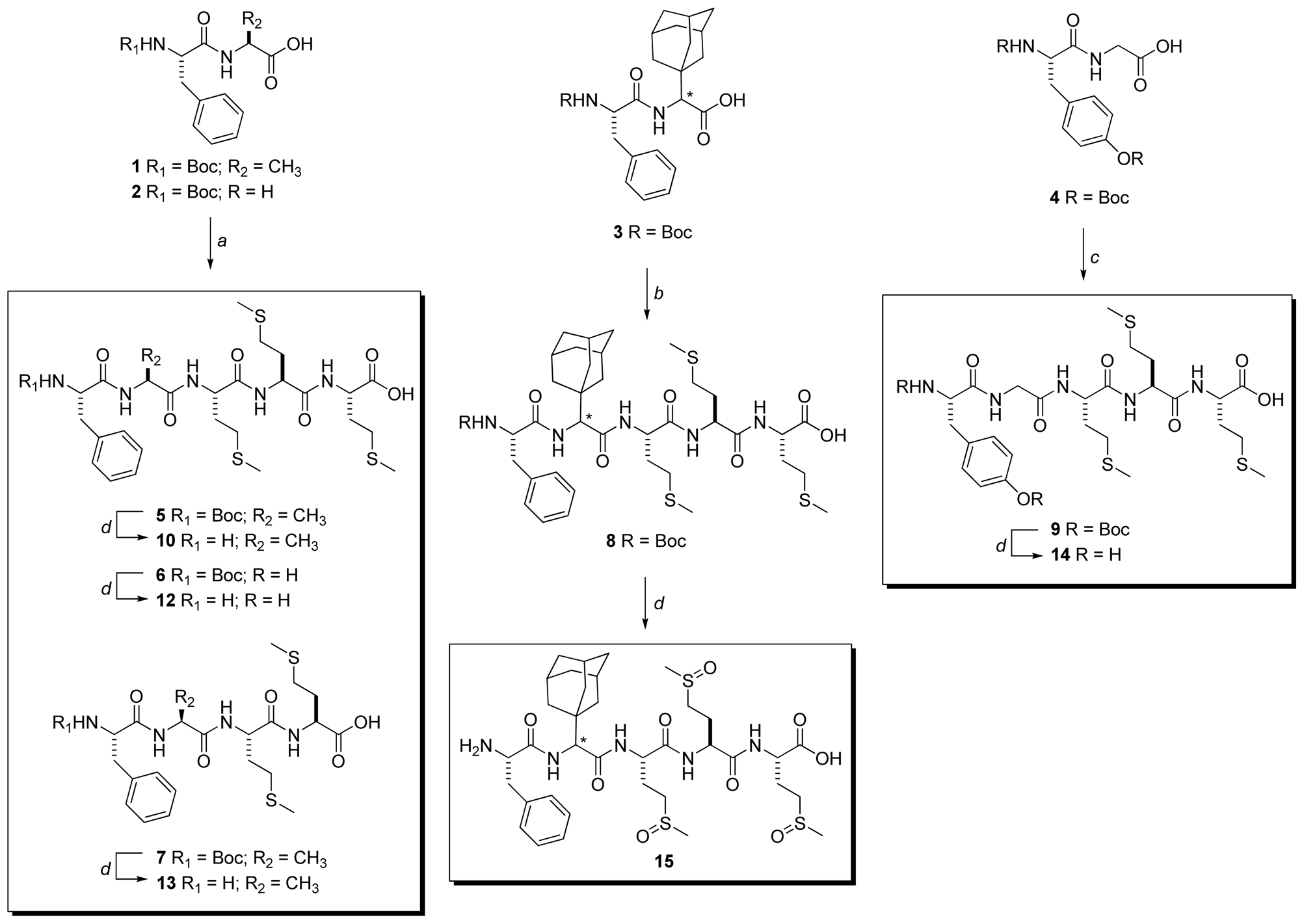
3.3.2. General Procedure for the Synthesis of Compounds 5–7
3.3.3. Boc-Phe-Ala-Met-Met-Met-OH (5)
3.3.4. Boc-Phe-Gly-Met-Met-Met-OH (6)
3.3.5. Boc-Phe-Ala-Met-Met-OH (7)
3.3.6. Synthesis of Boc-Phe-Ada-Met-Met-Met-OH (8)
3.3.7. Synthesis of Boc-Tyr(Boc)-Ala-Met-Met-Met-OH (9)
3.3.8. General procedure for the synthesis of compounds 10–14
3.3.9. H-Phe-Ala-Met-Met-Met-OH (10)
3.3.10. H-Phe-Gly-Met-Met-Met-OH (12)
3.3.11. H-Phe-Ala-Met-Met-OH (13)
3.3.12. H-Tyr-Ala-Met-Met-Met-OH (14)
3.3.13. H-Phe-Ada-Met(O)-Met(O)-Met(O)-OH (15)
3.4. Proliferation Assay
4. Conclusions
Supplementary Information
ijms-12-08415-s001.docxAcknowledgments
References
- Otvos, L., Jr. Peptide-based drug design: Here and now. Methods Mol. Biol. 2008, 494, 1–8. [Google Scholar]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than one reason to rethink the use of peptides in vaccine design. Nature Rev. Drug. Discov 2007, 6, 404–414. [Google Scholar]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer acetate in the treatment of multiple sclerosis emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar]
- Wilson, A.C.; Meethal, S.V.; Bowen, R.L.; Atwood, C.S. Leuprolide acetate: A drug of diverse clinical applications. Expert Opin. Investig. Drugs 2007, 16, 1851–1863. [Google Scholar]
- Wajcberg, E.; Tavaria, A. Exenatide: Clinical aspects of the first incretin-mimetic for the treatment of type 2 diabetes mellitus. Expert Opin. Pharmacother 2009, 10, 135–142. [Google Scholar]
- Zagon, I.S.; Verderame, M.F.; McLaughlin, P.J. The biology of the opioid growth factor receptor (OGFr). Brain Res. Rev 2002, 38, 351–376. [Google Scholar]
- Zagon, I.S.; Smith, J.P.; McLaughlin, P.J. Human pancreatic cancer cell proliferation in tissue culture is tonically inhibited by opioid growth factor. Int. J. Oncol 1999, 14, 577–584. [Google Scholar]
- Zagon, I.S.; Donahue, R.N.; Rogosnitzky, M.; McLaughlin, P.J. Imiquimod upregulates the opioid growth factor receptor to inhibit cell proliferation independent of immune function. Exp. Biol. Med. (Maywood) 2008, 8, 968–979. [Google Scholar]
- Smith, J.P.; Bingaman, S.I.; Mauger, D.T.; Harvey, H.H.; Demers, L.M.; Zagon, I.S. Opioid growth factor improves clinical benefit and survival in patients with advanced pancreatic cancer. Open Access J. Clin. Trials 2010, 2, 37–48. [Google Scholar]
- Zagon, I.S.; Jaglowski, J.R.; Verderame, M.F.; Smith, J.P.; Leure-Dupree, A.E.; McLaughlin, P.J. Combination chemotherapy with gemcitabine and biotherapy with opioid growth factor (OGF) enhances the growth inhibition of pancreatic adenocarcinoma. Cancer Chemother. Pharmacol 2005, 56, 510–520. [Google Scholar]
- Horvat, Š.; Mlinarić-Majerski, K.; Glavaš-Obrovac, Lj.; Jakas, A.; Veljković, J.; Marczi, S.; Kragol, G.; Roščić, M.; Matković, M.; Milostić-Srb, A. Tumor-cell targeted methionine-enkephalin analogues containing unnatural amino acids: Design, synthesis and in vitro antitumor activity. J. Med. Chem 2009, 49, 3136–3142. [Google Scholar]
- Gredičak, M.; Supek, F.; Kralj, M.; Majer, Z.; Hollosi, M.; Šmuc, T.; Mlinarić-Majerski, K.; Horvat, Š. Computational structure-activity study directs synthesis of novel antitumor enkephalin analogs. Amino Acids 2010, 38, 1185–1191. [Google Scholar]
- Cherkasov, A. Inductive QSAR descriptors. Distinguishing compounds with antibacterial activity by artificial neural networks. Int. J. Mol. Sci 2005, 6, 63–86. [Google Scholar]
- Fjell, D.C.; Jenssen, H.; Cheung, W.A.; Hancock, R.E.W.; Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and chemoinformatics. Chem. Biol. Drug Des 2011, 77, 48–56. [Google Scholar]
- Cherkasov, A.; Janković, B. Application of “inductive” QSAR descriptors for quantification of antibacterial activity of cationic polypeptides. Molecules 2004, 9, 1034–1052. [Google Scholar]
- Hastie, T.; Tibshirani, R.; Friedman, J. High-dimensional problems: p >> N. In The Elements of Statistical Learning: Data Mining, Inference and Prediction, 3rd ed.; Springer: Berlin, Germany, 2009; Volume Chapter 18, pp. 649–698. [Google Scholar]
- Braga-Neto, U.; Dougherty, E. Is cross-validation valid for small sample microarray classification? Bioinformatics 2004, 20, 374–380. [Google Scholar]
- Zhou, X.; Mao, K.Z. The ties problem resulting from counting-based error estimators and its impact on gene selection algorithms. Bioinformatics 2006, 22, 2507–2515. [Google Scholar]
- Chao, S.M.; Dougherty, E.R. The peaking phenomenon in the presence of feature-selection. Pat. Rec. Let 2008, 11, 1667–1674. [Google Scholar]
- Hua, J.; Xiong, Z.H.; Lowey, J.; Suh, E.; Dougherty, E.R. Optimal number of features as a function of sample size for various classification rules. Bioinformatics 2005, 21, 1509–1515. [Google Scholar]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. Royal. Statist. Soc. B 1996, 58, 267–288. [Google Scholar]
- Elad, M. Sparse and Redundant Representations—From Theory to Applications in Signal and Image Processingl; Springer: Berlin, Germany, 2010. [Google Scholar]
- Tropp, J.A.; Wright, S.J. Computational methods for sparse solution of linear inverse problems. Proc. IEEE 2010, 98, 948–958. [Google Scholar]
- Kim, S.J.; Koh, K.; Lustig, M.; Boyd, S.; Gorinevsky, S. An interior-point method for large-scale ℓ1 -regularized least squares. IEEE J. Sel. Top. Signal Proc 2007, 1, 606–617. [Google Scholar]
- Ein-Dor, L.; Zuk, O.; Domany, E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 5923–5928. [Google Scholar]
- Yang, F.; Mao, K.Z. Robust feature selection for microarray data based on multicriterion fusion. IEEE ACM Trans. Comp. Biol. Bioinforma 2011, 8, 1080–1092. [Google Scholar]
- Muselli, M.; Bertoni, A.; Frasca, M.; Beghini, A.; Ruffino, F.; Valentini, G. A mathematical model for the validation of gene selection methods. IEEE ACM Trans. Comp. Biol. Bioinforma 2011, 8, 1385–1392. [Google Scholar]
- Kavuk, O.D.; Kamada, M.; Akutsu, T.; Knapp, E.W. Prediction using step-wise L1, L2 regularization and feature selection for small data sets with large number of features. BMC Bioinforma 2011, 12, 412:1–412:10. [Google Scholar]
- A repository of structures, experimental data and QSAR models for molecules with antitumor activity, 2008. Anticancer.irb.hr Repository Web site. Available online: http://anticancer.irb.hr accessed on 18 November 2011.
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des 2005, 19, 453–463. [Google Scholar]
- Sadowski, J.; Gasteiger, J.; Klebe, G. Comparison of automatic three-dimensional model builders using 639 X-ray structures. J. Chem. Inf. Comput. Sci 1994, 34, 1000–1008. [Google Scholar]
- Marvin—Draw and visualize chemistry, 2008. ChemAxon Web site. Available online: http://www.chemaxon.com accessed on 18 November 2011.
- l1_ls: Simple Matlab Solver for l1-Regularized Least Square Problem, Beta Version; Stanford University: Stanford, CA, USA, 2001. Available online: http://www.stanford.edu/~boyd/l1_ls/ accessed on 18 November 2011.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank/(position on the lists/mean value) | Compounds | MW | log P | log S |
|---|---|---|---|---|
| 1. (1-1-1-1/1) | H-Phe-Ala-Met-Met-Met-OH (10) | 629.96 | 0.89 | −5.23 |
| 2. (2-3-2-4/2.75) | H-Phe-Ada-Met-Met-Met-OH (11) | 750.17 | 1.12 | −6.06 |
| 3. (5-2-4-3/3.25) | H-Phe-Gly-Met-Met-Met-OH (12) | 615.93 | 0.76 | −5.22 |
| 3. (4-4-3-2/3.25) | H-Phe-Ala-Met-Met-OH (13) | 498.74 | 0.62 | −4.70 |
| 5. (8-5-5-5/5.25) | H-Tyr-Ala-Met-Met-Met-OH (14) | 645.96 | 0.58 | −4.99 |
| Compounds below are listed on three lists only | ||||
| 6. (3-6-x -6/ 5) | H-Phe-Ada-Met-Met-OH | 618.95 | 0.91 | −5.70 |
| 7. (6-7-6 -x/ 6.33) | H-Tyr-Ada-Met-Met-Met-OH | 766.17 | 0.72 | −5.66 |
| Compounds below are listed on two lists only | ||||
| 8. (x-x-8-7/7.5) | H-Phe-Ala-Phe-Met-Met-OH | 645.93 | 0.78 | −5.42 |
| 9. (7-x-9-x/8) | H-Phe-Ada-Phe-Met-Met-OH | 766.14 | 1.40 | −5.77 |
| 10. (9-8-x-x/8.5) | H-Tyr-Ada-Met-Met-OH | 634.95 | 0.62 | −5.27 |
| 11. (x-x-7-10/8.5) | H-Phe-Ala-Met-Met-Gly-OH | 555.80 | 0.80 | −4.58 |
| 12. (x-10-x-8/9) | H-Phe-Gly-Met-Met-OH | 484.71 | 0.58 | −4.69 |
| Compounds below are listed on one list only | ||||
| 13. (x-9-x-x) | H-Tyr-Gly-Met-Met-Met-OH | 631.93 | 0.50 | −4.95 |
| 13. (10-x-x-x-x) | H-Phe-Ada-Met-Met-Gly-OH | 676.01 | 0.83 | −5.53 |
| 13.(x-x-x-9) | H-Tyr-Ala-Met-Met-OH | 514.74 | 0.51 | −4.26 |
| 13. (x-x-10-x) | H-Tyr-Ala-Met-Met-OH | 514.74 | 0.51 | −4.26 |
| Top-rated compounds predicted in [12] | ||||
| H-Tyr-Ada-Gly-Phe-Met-OH |  | - | - | - |
| H-Phe-Ada-Gly-Phe-Met-OH | ||||
| H-Phe-Ada-Gly-Phe-Phe-OH | ||||
| H-Tyr-Ada-Gly-Phe-Phe-OH | ||||
| H-Trp-Ada-Gly-Phe-Met-OH | ||||
| H-Tyr-Ada-Gly-Phe-Gly-OH | ||||
| H-Trp-Ada-Gly-Phe-Phe-OH | ||||
| H-Phe-Ada-Gly-OH | ||||
| H-Phe-Gly-Gly-Phe-Phe-OH | ||||
| H-Phe-Gly-Aaa-Gly-OH | ||||
| sparse | Lin SVM | MLP | RBF SVM | Poly SVM | KNN | |
|---|---|---|---|---|---|---|
| sparse | 1.0 | 0.9701 | 0.9391 | 0.9406 | 0.5510 | 0.3275 |
| Lin SVM | - | 1.0 | 0.9631 | 0.9568 | 0.4860 | 0.3141 |
| MLP | - | - | 1.0 | 0.9359 | 0.4152 | 0.2685 |
| RBF SVM | - | - | - | 1.0 | 0.6278 | 0.3631 |
| Poly SVM | - | - | - | - | 1.0 | 0.1396 |
| KNN | - | - | - | - | - | 1.0 |
| Compound | PG(%) a | ||
|---|---|---|---|
| SW620 | MCF-7 | HeLa | |
| H-Phe-Ala-Met-Met-Met-OH (10) | 100 | 74 | 88 |
| H-Phe-Gly-Met-Met-Met-OH (12) | 95 | 86 | 86 |
| H-Phe-Ala-Met-Met-OH (13) | 86 | 68 | 71 |
| H-Tyr-Ala-Met-Met-Met-OH (14) | 97 | 71 | 91 |
| H-Phe-Ada-Met(O)-Met(O)-Met(O)-OH (15) | 77 | 82 | 64 |
| H-Tyr-Gly-Gly-Phe-Met-OH (OGF) | 85 | 92 | 88 |
| H-Phe-Ada-Gly-Phe-Met-OH b | 35 | 39 | 49 |
| H-Phe-Ada-Gly-Phe-Phe-OH b | 39 | 41 | 23 |
| H-Tyr-Ada-Gly-Phe-Phe-OH b | 38 | 81 | 71 |
| H-Tyr-(S)-Ada-OH c | 72 | 100 | 92 |
| H-Tyr-(R)-Ada-OH c | 88 | 99 | 97 |
| H-Tyr-(S)-Ada-Gly-OH c | 85 | 100 | 90 |
| H-Tyr-(R)-Ada-Gly-OH c | 87 | 100 | 89 |
| H-Tyr-Ada-Gly-Phe-Met-OH c | 85 | 100 | 85 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radman, A.; Gredičak, M.; Kopriva, I.; Jerić, I. Predicting Antitumor Activity of Peptides by Consensus of Regression Models Trained on a Small Data Sample. Int. J. Mol. Sci. 2011, 12, 8415-8430. https://doi.org/10.3390/ijms12128415
Radman A, Gredičak M, Kopriva I, Jerić I. Predicting Antitumor Activity of Peptides by Consensus of Regression Models Trained on a Small Data Sample. International Journal of Molecular Sciences. 2011; 12(12):8415-8430. https://doi.org/10.3390/ijms12128415
Chicago/Turabian StyleRadman, Andreja, Matija Gredičak, Ivica Kopriva, and Ivanka Jerić. 2011. "Predicting Antitumor Activity of Peptides by Consensus of Regression Models Trained on a Small Data Sample" International Journal of Molecular Sciences 12, no. 12: 8415-8430. https://doi.org/10.3390/ijms12128415