Validation of Quantitative Structure-Activity Relationship (QSAR) Model for Photosensitizer Activity Prediction

Abstract
:1. Introduction
2. Results and Discussion
2.1. QSAR Modeling
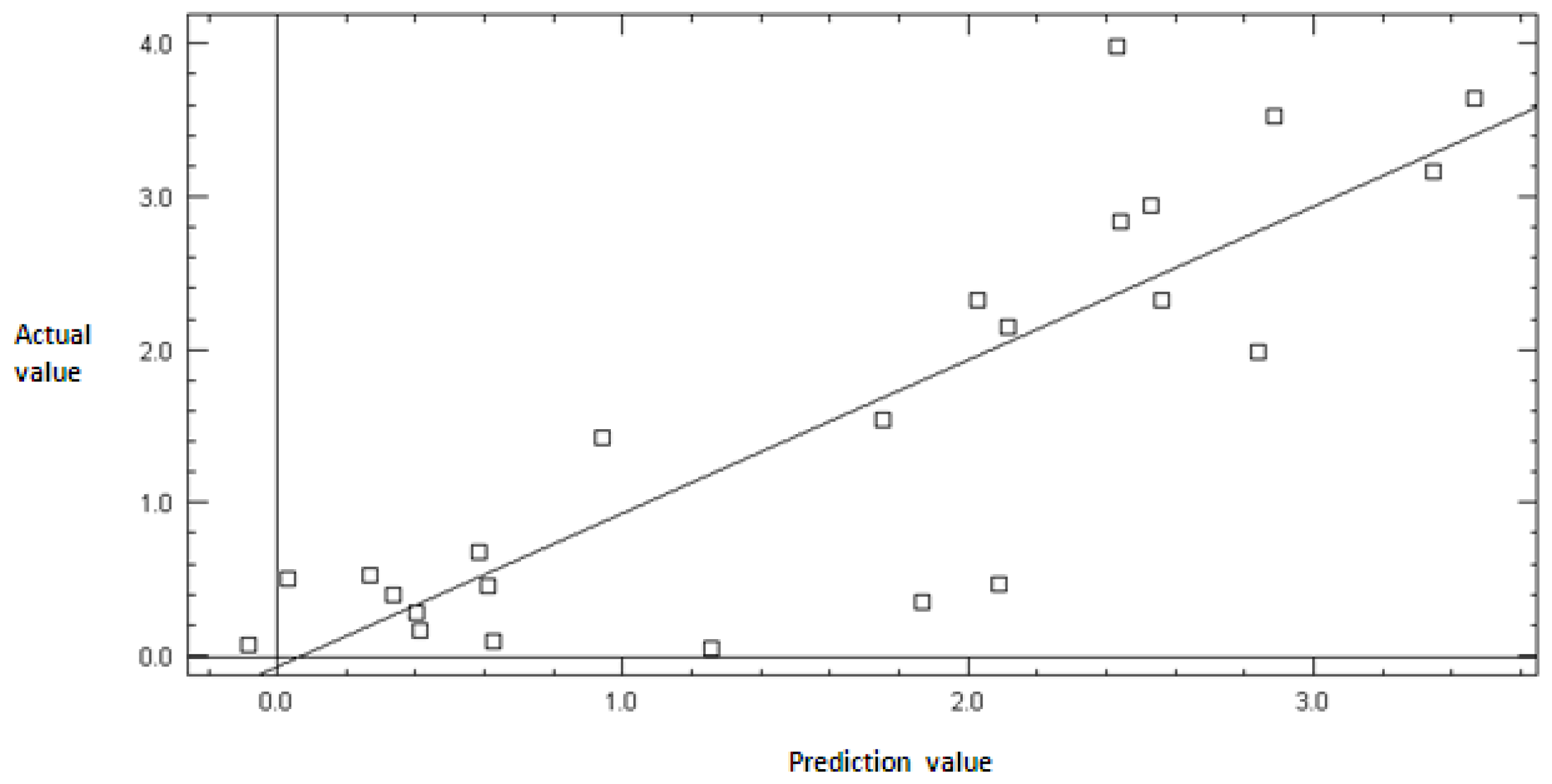
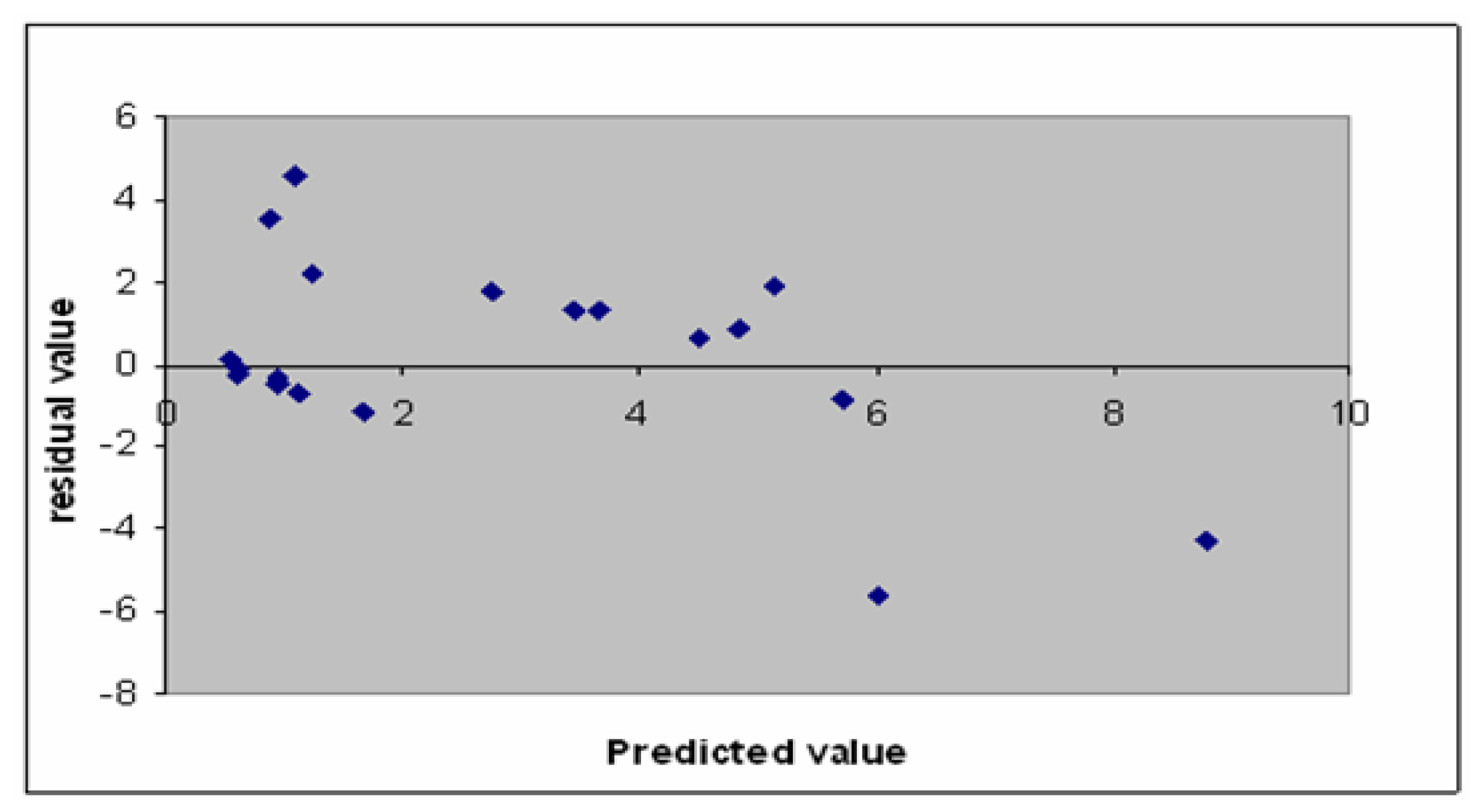
2.2. Model Validation
3. Experimental
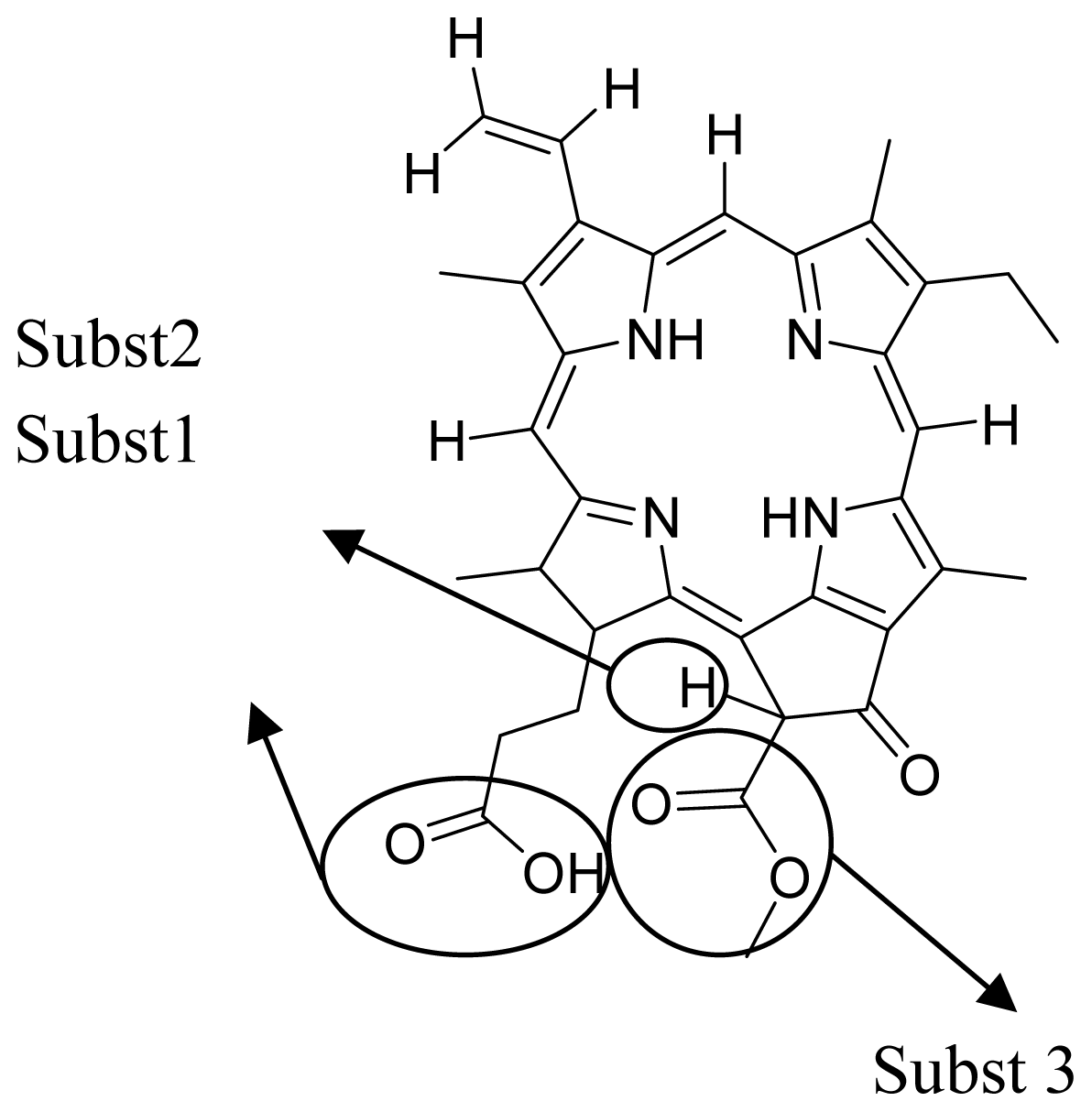
3.1. QSAR Modeling of Porphyrin
3.2. Development of QSAR Model
3.3. Model Validation
3.4. Preparation of Compounds for External Test Set
3.5. Determination of Photocytotoxicity of Compounds in External Test Set by MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium hydrobromide) Assay
4. Conclusions
Acknowledgements
References
- Gerard, L.C.C.; Halimah, Y. Second Report of National Cancer Register, Cancer Incidence in Malaysia; National Cancer Registry: Kuala Lumpur, Malaysia, 2003. [Google Scholar]
- Nyman, E.; Hynninen, P.H. Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B 2004, 73, 1–28. [Google Scholar]
- Henderson, B.W.; Dougherty, T.J. How does photodynamic therapy works. J. Photochem. Photobiol 1992, 55, 145–157. [Google Scholar]
- Detty, M.R.; Gibson, S.L.; Wagner, S.J. Current clinical and preclinical photosensitizers for use in photodynamic therapy. J. Med. Chem 2004, 47, 3897–3915. [Google Scholar]
- Dougherty, T.J. Photosensitizers: Therapy and detection of malignant tumours. J. Photochem. Photobiol 1987, 45, 879–889. [Google Scholar]
- Dougherty, T.J. Photodynamic therapy. J. Photochem. Photobiol 1993, 58, 895–900. [Google Scholar]
- Macdonald, I.D.T. Basic principles of photodynamic therapy. J. Porphyr. Phthalocya 2001, 5, 105–129. [Google Scholar]
- Bonnett, R. Chemical Aspects of Photodynamic Therapy; Gordon and Breach Science: Amsterdam, The Netherlands, 2000. [Google Scholar]
- Allison, R.R.; Downie, G.H.; Cuenca, R.; Hu, X.H.; Child, C.J.; Sibata, C.H. Photosensitizers in clinical PDT. Photodiagnosis Photodyn. Ther 2004, 1, 27–42. [Google Scholar]
- Vanyur, R.; Heberger, K.; Kovesdi, I.; Jakus, J. Prediction of tumoricidal activity and accumulation of photosensitizers in photodynamic therapy using multiple linear regression and artificial neural networks. J. Photochem. Photobiol 2002, 75, 471–478. [Google Scholar]
- Boyle, R.W.; Dolphyn, D. Structure and biodistribution relationships of photodynamic sensitizers. J. Photochem. Photobiol 1996, 64, 469–485. [Google Scholar]
- Henderson, B.W.; Bellnier, D.A.; Greco, W.R.; Sharma, A.; Pandey, R.K.; Vanghan, L.A.; Weishaupt, K.R.; Dougherty, T.J. An in vivo quantitative structure-activity relationship for congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res 1997, 57, 4000–4007. [Google Scholar]
- Potter, W.R.; Henderson, B.W.; Bellnier, D.A.; Pandey, R.K.; Vanghan, L.A.; Weishaupt, K.R.; Douherty, T.J. Parabolic quantitative structure-activity and photodynamic therapy: Application of three compartment model with clearance to the in vivo quantitative structure-activity relationships of a congeneric series of pyropheoporbide derivatives used as photosensitizers for photodynamic therapy. J. Photochem. Photobiol 1999, 70, 781–788. [Google Scholar]
- Depnath, A.K.; Jiang, S.; Strick, N.; Lin, K.; Haberfield, P.; Neurath, A.R. Three-dimensional structure-activity analysis of a series of porphyrins derivatives with anti hiv-1 activity targeted to the v3 loop of the gp120 envelope glycoprotein of the human immunodeficiency virus type 1. J. Med. Chem 1994, 37, 1099–1108. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Predictive qsar modeling diversity sampling of experimental data set and test set selection. J. Comput. Aided Mol. Des 2002, 5, 231–243. [Google Scholar]
- Beebe, K.R.; Pell, R.J.; Seasholtz, M.B. Chemometrics, a Practical Guide; Wiley Interscience: New York, NY, USA, 1998. [Google Scholar]
- Li, H.; Sun, J.; Sui, X.; Liu, J.; Yan, Z.; Liu, X.; Sun, Y.; He, Z. First-principle, structure-based prediction of hepatic metabolic clearance values in human. Eur. J. Med. Chem 2009, 44, 1600–1606. [Google Scholar]
- Vicini, P.; Geronikaki, A.; Incerti, M.; Zani, F.; Dearden, J.; Hewitt, M. 2-heteroarylimino-5- benzylidene-4-thiazolidinones analogues of 2-thiazolylimino-5-benzylidene-4-thiazolidinones with antimicrobial activity: Synthesis and structure—activity relationship. Bioorg. Med. Chem 2008, 16, 3714–3724. [Google Scholar]
- Korhonen, S.M. A fuzzy superposition and qsar technique towards an automated computational detection of biologically active compounds using multivariate methods. PhD Thesis, University of Kuopio, Kuopio, Finland, 2007. [Google Scholar]
- Lin, B.; Pirrung, M.C.; Deng, L.; Li, Z.; Liu, Y.; Webster, N.J.G. Neuroprotection by small molecule activators of the nerve growth factor receptor. J. Pharmacol. Exp. Ther 2007, 322, 59–69. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model 2002, 20, 269–276. [Google Scholar]
- Windows Reference Guide, TSAR version 3.3; Oxford Molecular, Ltd.: Oxford, UK, 2000.
- Banfi, S.; Caruso, E.; Buccafurni, L.; Murano, R.; Monti, E.; Gariboldi, M.; Papa, E.; Gramatica, P. Comparison between 5,10,15,20-tetraaryl- and diarylporphyrins as photosensitizers: Synthesis, photodynamic activity and quantitative structure activity relationship modeling. J. Med. Chem 2006, 49, 3293–3304. [Google Scholar]
- Banfi, S.; Caruso, E.; Caprioli, S.; Mazzagatti, L.; Canti, G.; Ravizza, R.; Gariboldi, M.; Monti, E. Photodynamic effects of porphyrin and chlorin photosensitizers in human colon adenocarcinoma cells. Bioorg. Med. Chem. 2004, 14, 4853–4860. [Google Scholar]
- ChemDraw, version 6.0; Cambridge Scientific Computing, Inc.: Cambridge, MA, USA, 2004.
- TSAR, version 3.3; Oxford Molecular, Ltd.: Oxford, UK, 2000.
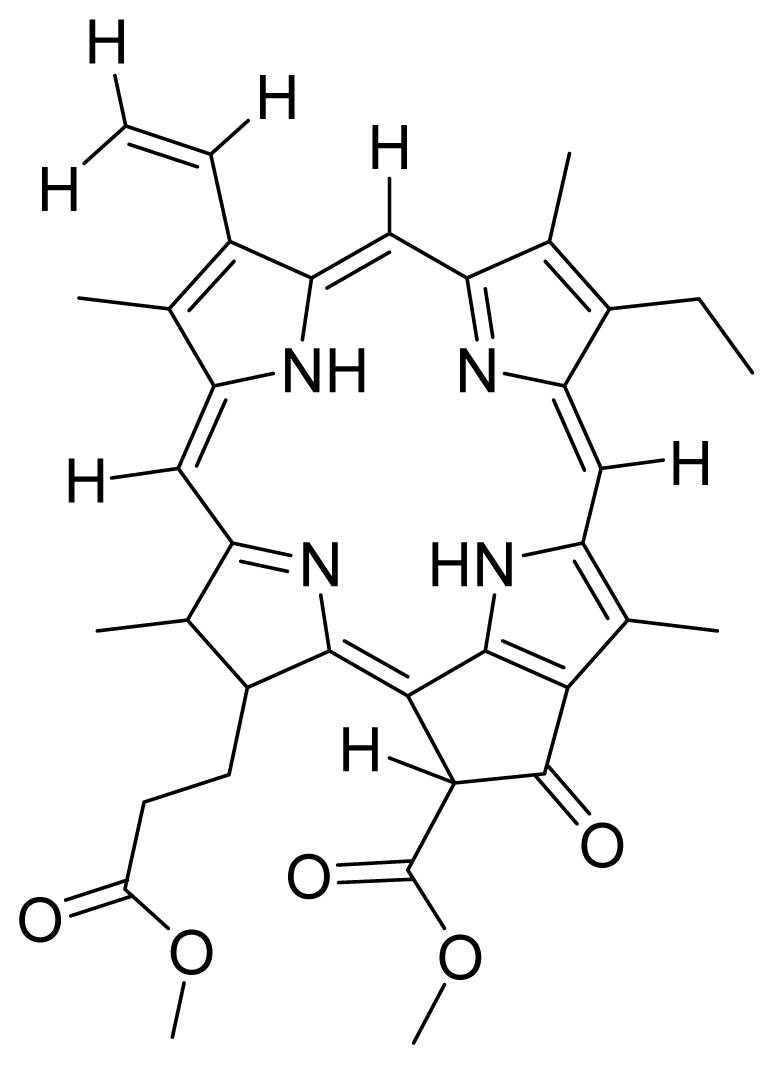
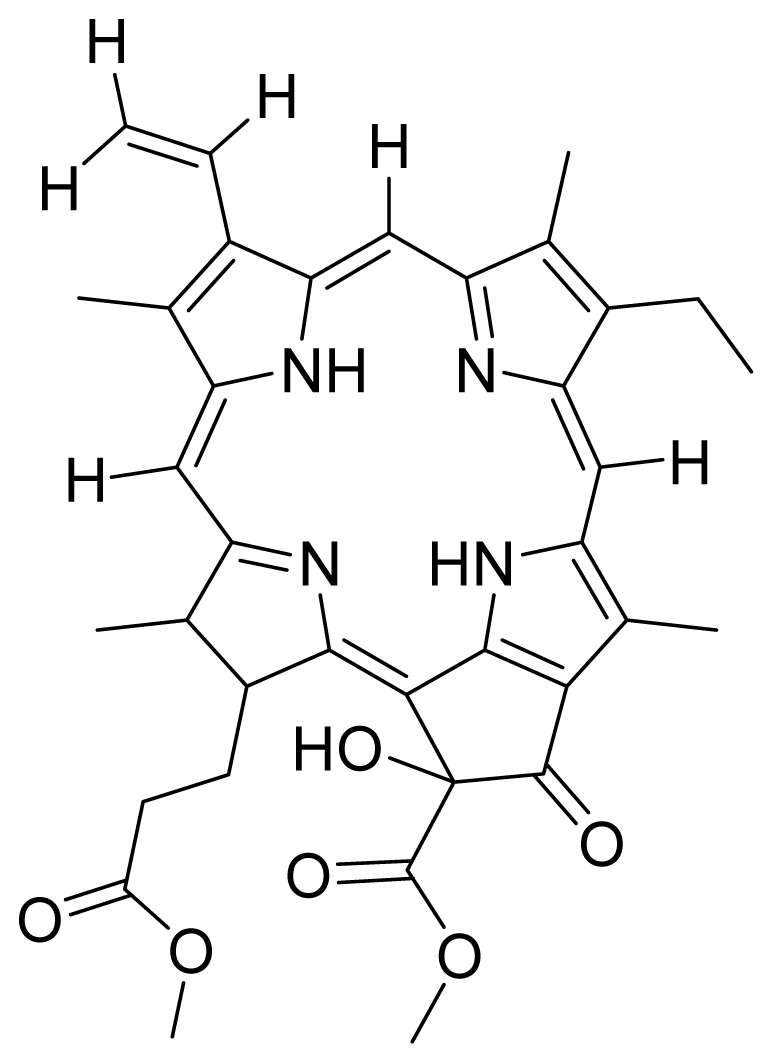
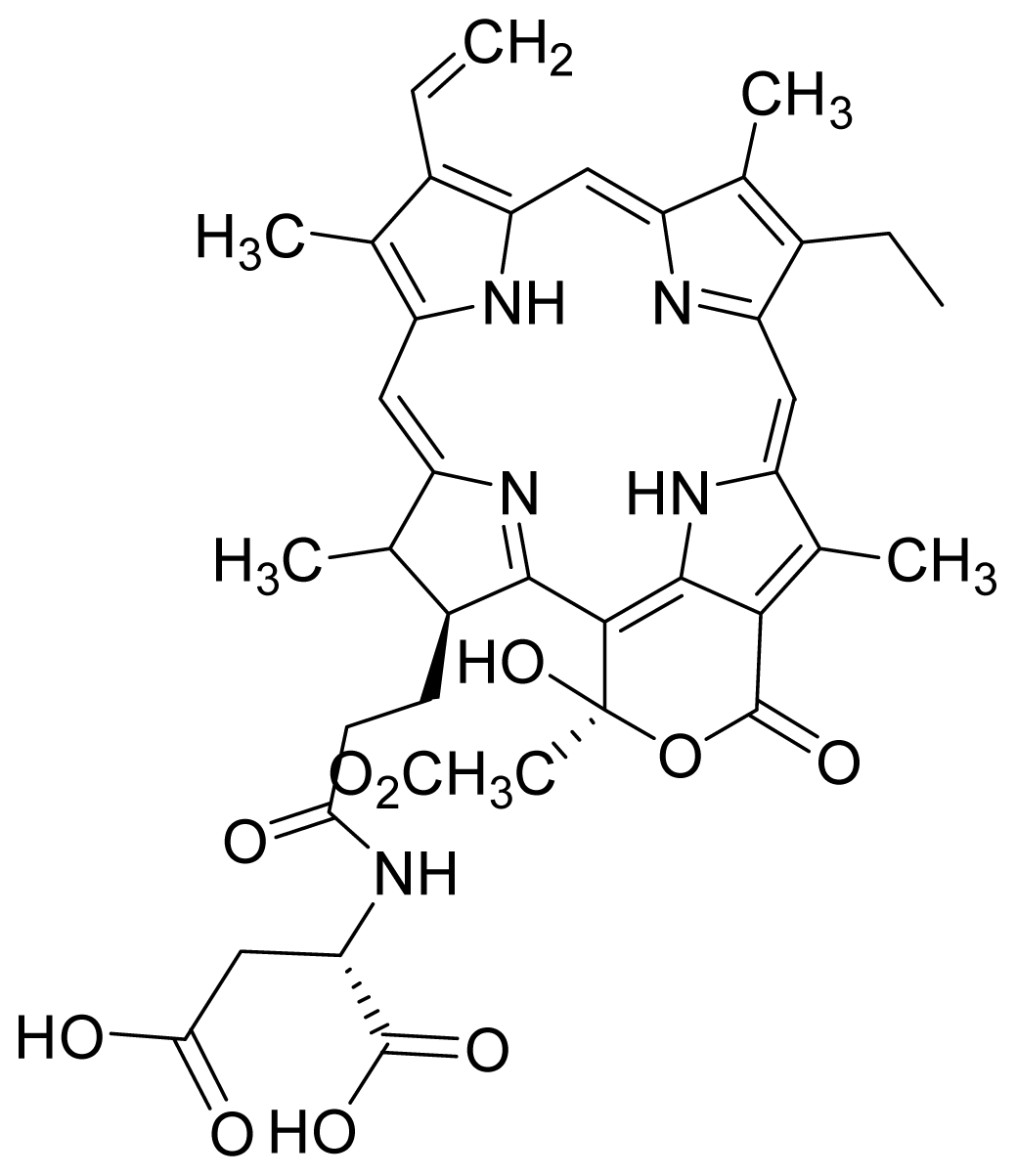
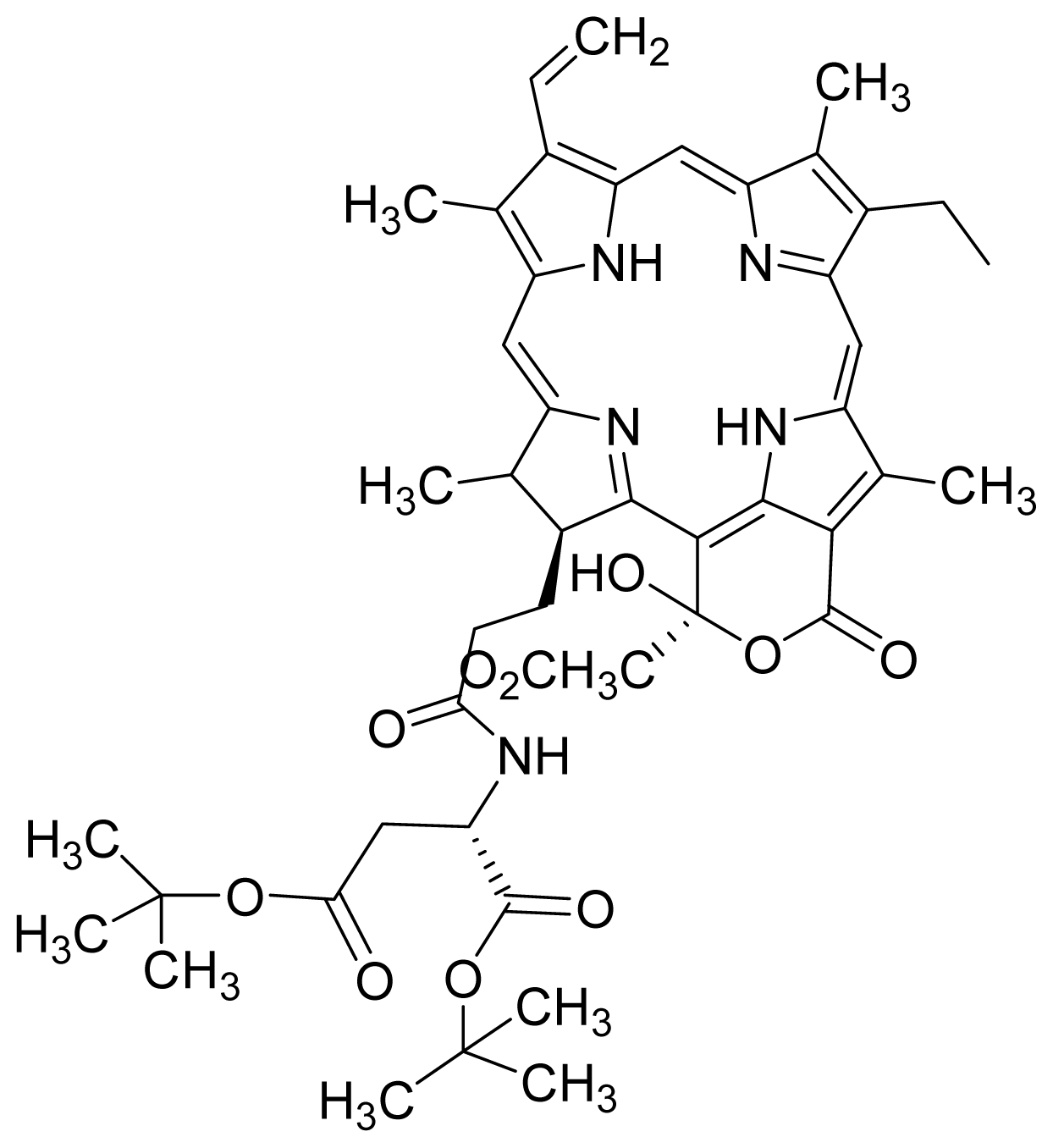
- Kamarulzaman, F.A.; Shaari, K.; Ho, A.S.H.; Lajis, N.H.; Teo, S.H.; Lee, H.B. Derivatives of pheophorbide-a and pheophorbide-b from photocytotoxic piper penangense extract. Chem. Biodivers 2011, 8, 494–502. [Google Scholar]
- Tan, P.J.; Appleton, D.R.; Mustafa, M.R.; Lee, H.B. Rapid identification of cyclic tetrapyrrolic photosensitisers for photodynamic therapy using on-line hyphenated lc-pda-ms coupled with photocytotoxicity assay. Phytochem. Anal 2011. [Google Scholar] [CrossRef]
- Harris, P.G.; Pearce, G.E.S.; Peakman, T.M.; Maxwell, J.R. A widespread and abundant chlorophyll transformation product in aquatic environments. Org. Geochem 1995, 23, 183–187. [Google Scholar]
- Demberelnyamba, D.; Ariuna, M.; Shim, Y.K. Newly synthesized water-soluble cholinium-purpurin photosensitizers and their stabilized gold nanoparticle as promising anticancer agents. Int. J. Mol. Sci 2008, 9, 864–871. [Google Scholar]
- Ocampo, R.; Repeta, D.J. Structural determination of purpurin-18 (as methyl ester) from sedimentary organic matter. Org. Geochem 1999, 30, 189–193. [Google Scholar]
- Wongsinkongman, P.; Brossi, A.; Wang, H.K.; Bastow, K.F.; Lee, K.H. Antitumor agents. Part 209: Pheophorbide-a derivatives as photoindependent cytotoxic agents. Bioorg. Med. Chem 2002, 10, 583–591. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Statistical Output | Value |
|---|---|
| Non-cross validated r2 | 0.87 |
| Cross validation r2 (CV) | 0.71 |
| F-value | 37.85 |
| F-probability | 1.95 × 10−8 |
| Standard error of estimate (SEE) | 0.49 |
| Residual sum of square (RSS) | 4.12 |
| Predictive sum of square (PRESS) | 9.23 |
| No. | Verloop B2 (subst. 1) | Inert 3 Length | Vamp Octupole ZZY | Exp IC50 (μM) |
|---|---|---|---|---|
| 1 | 0.00 | 0.02 | 0.49 | 0.39 |
| 2 | 0.17 | 0.03 | 0.85 | 0.52 |
| 3 | 0.00 | 0.11 | 0.52 | 0.51 |
| 4 | 0.17 | 0.11 | 0.44 | 0.39 |
| 5 | 0.79 | 0.71 | 0.75 | 0.68 |
| 6 | 1.00 | 1.00 | 1.00 | 0.50 |
| 7 | 0.00 | 0.13 | 0.63 | 0.45 |
| 8 | 0.00 | 0.13 | 0.73 | 5.63 |
| 9 | 0.00 | 0.14 | 0.67 | 0.44 |
| 10 | 1.00 | 0.98 | 0.77 | 5.69 |
| 11 | 0.00 | 0.00 | 0.18 | 4.47 |
| 12 | 0.17 | 0.29 | 0.93 | 4.96 |
| 13 | 0.84 | 0.84 | 0.99 | 7.04 |
| 14 | 0.00 | 0.00 | 0.56 | 0.62 |
| 15 | 0.90 | 0.12 | 0.74 | 4.86 |
| 16 | 0.00 | 0.09 | 0.58 | 4.45 |
| 17 | 0.89 | 0.14 | 0.15 | 4.72 |
| 18 | 0.80 | 0.14 | 0.29 | 3.43 |
| 19 | 0.92 | 0.15 | 0.00 | 5.11 |
| 20 | 0.97 | 0.17 | 0.30 | 4.49 |
| Descriptors | Regression Coefficient a | Jacknife SE b | Covariance SE c | t-Value d | t-Probability e |
|---|---|---|---|---|---|
| Verloop B2 | 0.96 | 0.41 | 0.44 | 2.16 | 0.05 |
| Inertia moment 3 length | 6.42 | 0.52 | 0.73 | 8.75 | 1.04 × 10−7 |
| Vamp octupole ZZY | −1.63 | 1.06 | 0.80 | −2.03 | 0.06 |
| Descriptor | Symbol | Explanation |
|---|---|---|
| Verloop parameter | Verloop B2 (substituent 1) | The distance from the axis of the attachment bond, measured perpendicularly to the edge of the substituents. |
| Molecular attributes | Inertia moment 3 length | Indicates the strength and orientation behaviors of molecule in an electrostatic field. |
| Electrostatic parameter | Vamp octupole ZZY | Properties of molecule arising from the interaction between a charge probe, such as positive unit point reflecting a proton, and target molecule. |
| Compounds No. | Experimental log 1/IC50 | Predicted log 1/IC50 |
|---|---|---|
| 1 | 1.39 | 1.70 |
| 2 | 1.39 | 1.83 |
| 3 | 1.37 | 2.12 |
| 4 | 1.25 | 2.12 |
| 5 | 1.11 | 1.58 |
| 6 | 1.03 | 1.84 |
| 7 | 0.97 | 1.54 |
| 8 | 0.96 | 1.46 |
| 9 | 0.82 | 1.66 |
| 10 | 0.80 | 1.32 |
| 11 | 0.78 | 1.42 |
| 12 | 0.71 | 1.72 |
| No. | Compounds | Exp. Value (μM) | Pred. Value (μM) | No. | Compounds | Exp. Value (μM) | Pred. Value (μM) |
|---|---|---|---|---|---|---|---|
| 1 |  Pheophorbide A (pha) | 0.39 | 6.02 | 2 |  Pyropheophorbide A | 0.52 | 0.6 |
| 3 |  Pheophorbide A methyl ester | 0.51 | 1.65 | 4 |  Hydroxy pheophorbide A methyl ester | 0.39 | 0.61 |
| 5 |  G2 aspartyl (deprotected) | 0.68 | 0.54 | 6 |  G2 aspartyl (protected) | 0.50 | 0.56 |
| 7 |  Hydroxy pheophorbide B methyl ester | 0.45 | 1.12 | 8 |  Methoxy G2 methyl ester (a type) | 5.63 | 1.08 |
| 9 |  G2 dimethyl ester (151- hydroxypurpurin-7-lactone methyl diester) | 0.44 | 0.95 | 10 |  Purpurin 18 (KMP1) | 5.69 | 4.82 |
| 11 |  Purpurin-18 methyl ester | 4.47 | 8.78 | 12 |  G2 acid methyl (151-hydroxypurpurin-7-lactone methyl ester) | 4.96 | 3.67 |
| 13 |  Chlorophyllone a | 0.62 | 0.95 | 14 |  G2 lysine (protected) | 7.04 | 5.15 |
| 15 |  Hydroxy pheophorbide A | 4.45 | 0.88 | 16 |  G2 lysine deprotected | 4.86 | 5.72 |
| 17 |  Purpurin Lys | 4.72 | 3.43 | 18 |  Pha Asp | 3.43 | 1.23 |
| 19 |  Pha Lys | 5.11 | 4.49 | 20 |  HO-Pha-Lys | 4.49 | 2.76 |
| Descriptor | Statistics
| Descriptor | Statistics
| ||
|---|---|---|---|---|---|
| X̄ | SD | X̄ | SD | ||
| Verloop L (subst. 1) | 0.50 | 0.29 | Verloop L (subst. 2) | 0.23 | 0.26 |
| Verloop B1 (subst. 1) | 0.54 | 0.26 | Verloop B1 (subst. 2) | 0.66 | 0.32 |
| Verloop B2 (subst. 1) | 0.37 | 0.25 | Verloop B2 (subst. 3) | 0.05 | 0.11 |
| Verloop B4 (subst. 1) | 0.58 | 0.28 | Verloop B5 (subst. 2) | 0.48 | 0.31 |
| Inert. Moment 2 size | 0.14 | 0.07 | Inert. Moment 1length | 0.31 | 0.27 |
| Inert. Moment 3 length | 0.23 | 0.15 | Ellipsoidal volume | 0.15 | 0.07 |
| Log P | 0.60 | 0.26 | Total lipole | 0.37 | 0.25 |
| Lipole X component | 0.34 | 0.22 | Lipole Z component | 0.53 | 0.27 |
| Kier ChiV5 (ring) | 0.24 | 0.33 | Kappa 2 | 0.22 | 0.15 |
| Balaban topological | 0.42 | 0.29 | ADME H bond donor | 0.14 | 0.28 |
| ADME violation | 0.27 | 0.27 | VAMP total energy | 0.78 | 0.15 |
| VAMP heat of formation | 0.68 | 0.18 | VAMP HOMO | 0.44 | 0.14 |
| VAMP polarization XX | 0.26 | 0.13 | VAMP polarization XY | 0.41 | 0.29 |
| VAMP polarization XZ | 0.49 | 0.25 | VAMP polarization YY | 0.33 | 0.16 |
| VAMP polarization YZ | 0.47 | 0.25 | VAMP polarization ZZ | 0.33 | 0.24 |
| VAMP quadpole XX | 0.62 | 0.16 | VAMP quadpole XY | 0.57 | 0.19 |
| VAMP quadpole XZ | 0.58 | 0.26 | VAMP quadpole YY | 0.55 | 0.18 |
| VAMP quadpole YZ | 0.34 | 0.21 | VAMP quadpole ZZ | 0.25 | 0.10 |
| VAMP octupole XXX | 0.14 | 0.09 | VAMP octupole XXY | 0.88 | 0.07 |
| VAMP octupole XXZ | 0.27 | 0.11 | VAMP octupole YYX | 0.88 | 0.10 |
| VAMP octupole YYY | 0.42 | 0.23 | VAMP octupole YYZ | 0.89 | 0.07 |
| VAMP octupole ZZX | 0.75 | 0.19 | VAMP octupole ZZY | 0.59 | 0.23 |
| VAMP octupole ZZZ | 0.57 | 0.19 | VAMP octupole XYZ | 0.19 | 0.08 |
| Total dipole | 0.26 | 0.16 | Dipole x component | 0.19 | 0.13 |
| Dipole Y component | 0.52 | 0.27 | Dipole Z component | 0.60 | 0.23 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Frimayanti, N.; Yam, M.L.; Lee, H.B.; Othman, R.; Zain, S.M.; Rahman, N.A. Validation of Quantitative Structure-Activity Relationship (QSAR) Model for Photosensitizer Activity Prediction. Int. J. Mol. Sci. 2011, 12, 8626-8644. https://doi.org/10.3390/ijms12128626
Frimayanti N, Yam ML, Lee HB, Othman R, Zain SM, Rahman NA. Validation of Quantitative Structure-Activity Relationship (QSAR) Model for Photosensitizer Activity Prediction. International Journal of Molecular Sciences. 2011; 12(12):8626-8644. https://doi.org/10.3390/ijms12128626
Chicago/Turabian StyleFrimayanti, Neni, Mun Li Yam, Hong Boon Lee, Rozana Othman, Sharifuddin M. Zain, and Noorsaadah Abd. Rahman. 2011. "Validation of Quantitative Structure-Activity Relationship (QSAR) Model for Photosensitizer Activity Prediction" International Journal of Molecular Sciences 12, no. 12: 8626-8644. https://doi.org/10.3390/ijms12128626