Combined Pharmacophore Modeling, Docking, and 3D-QSAR Studies of PLK1 Inhibitors

Abstract
:1. Introduction
2. Results and Discussion
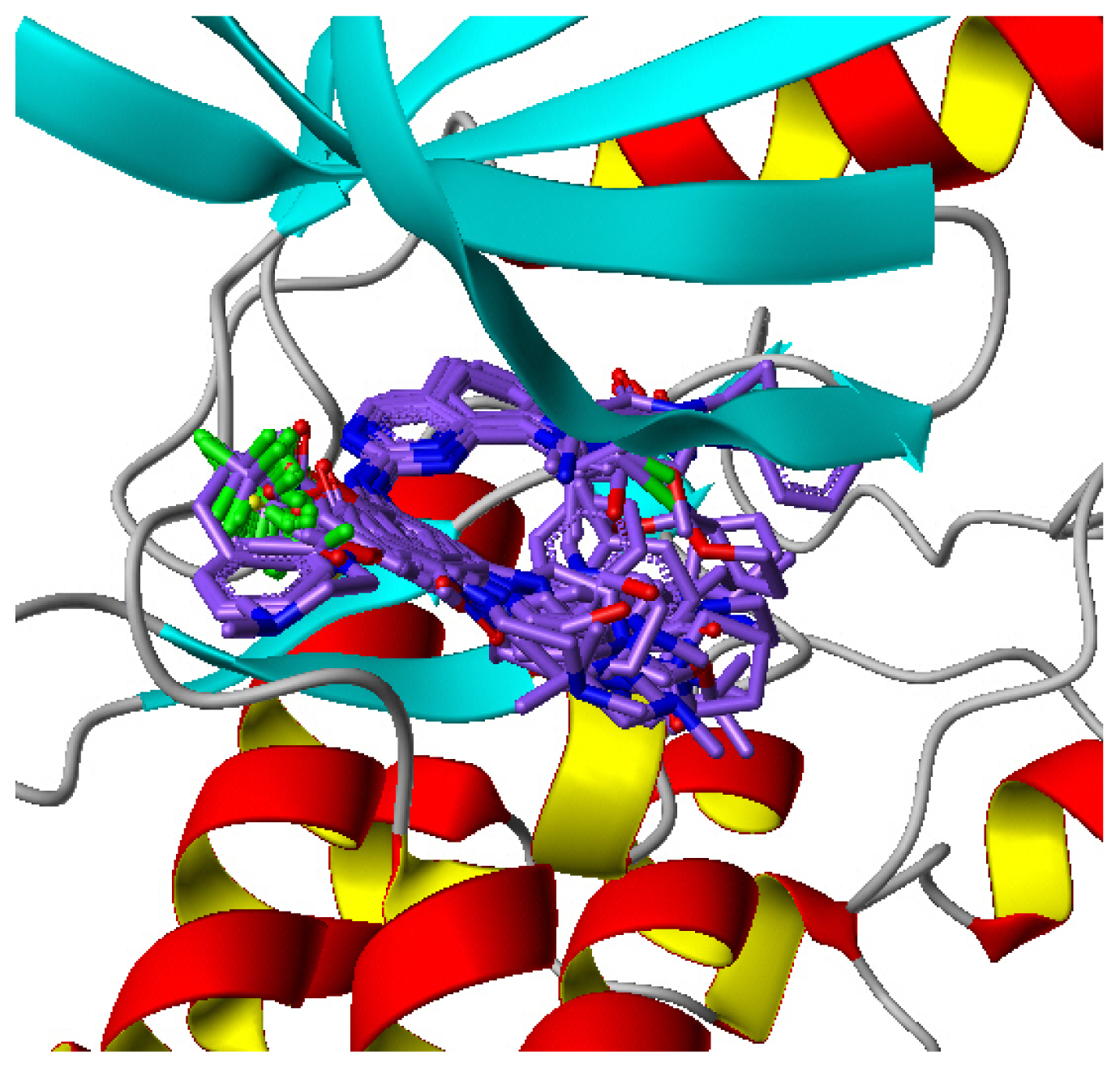
2.1. Multiple Structure Alignment Analyses
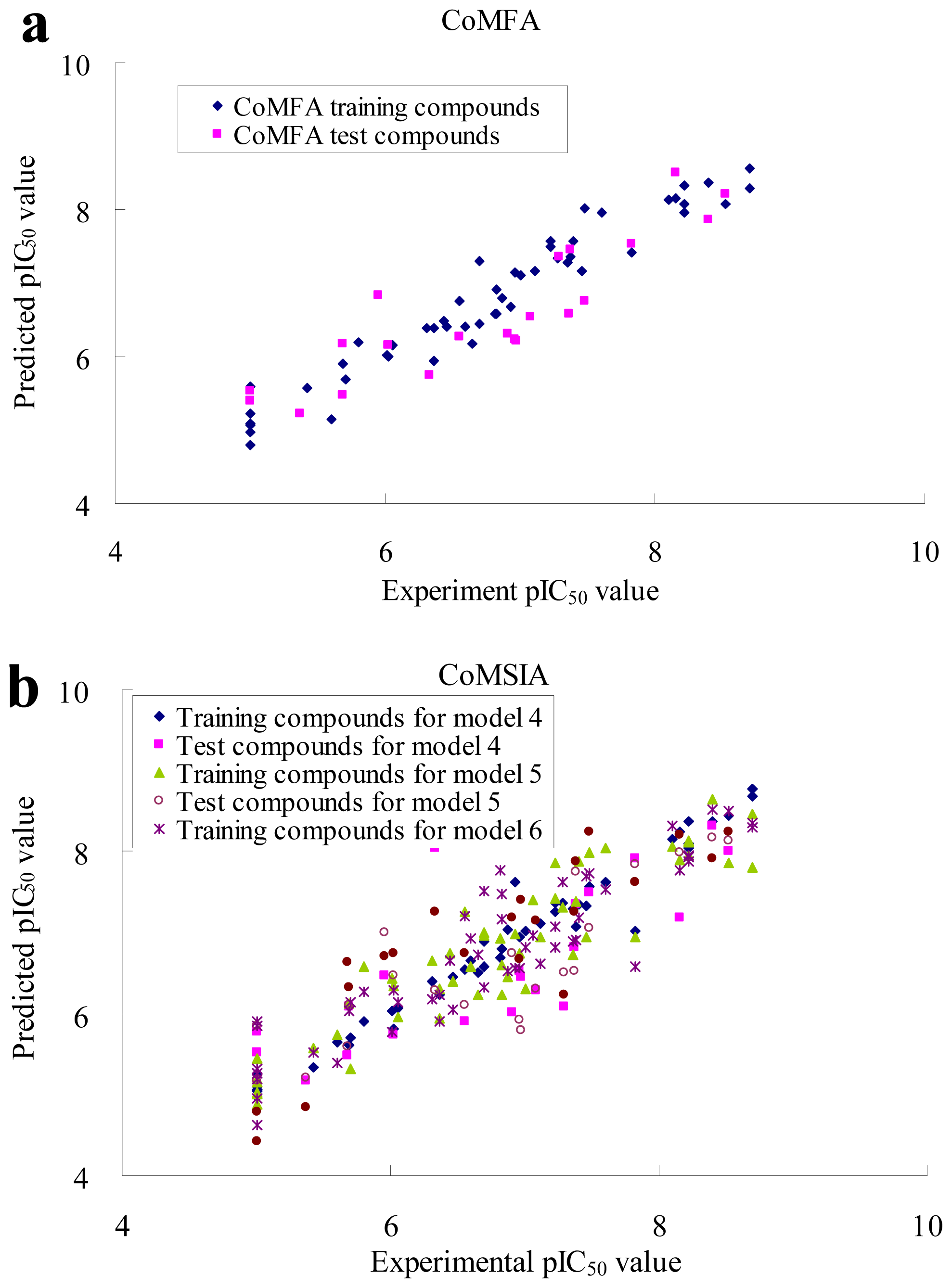
2.2. CoMFA and CoMSIA Statistical Results
2.3. CoMFA Contour Maps
2.4. CoMSIA Contour Maps
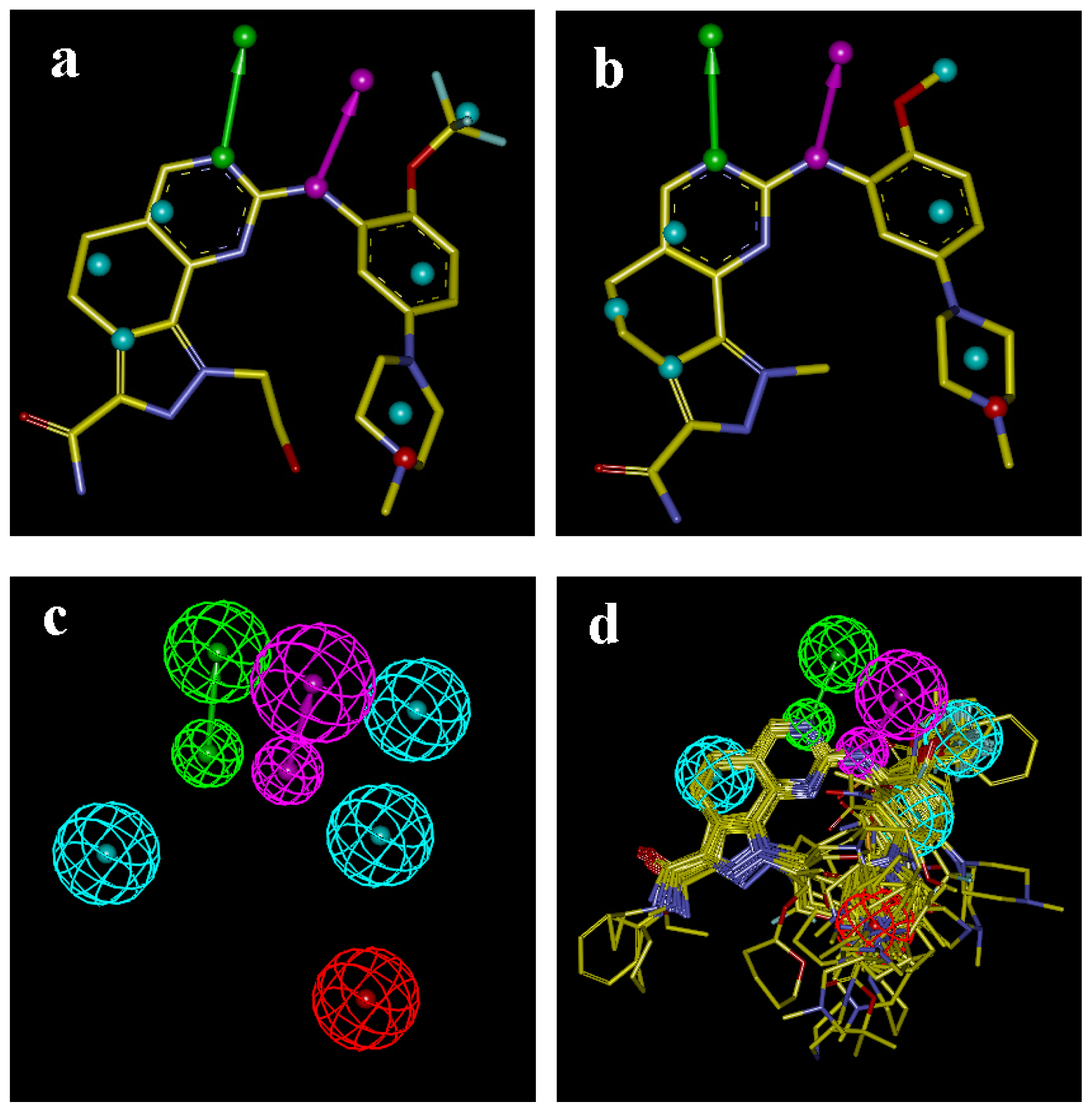
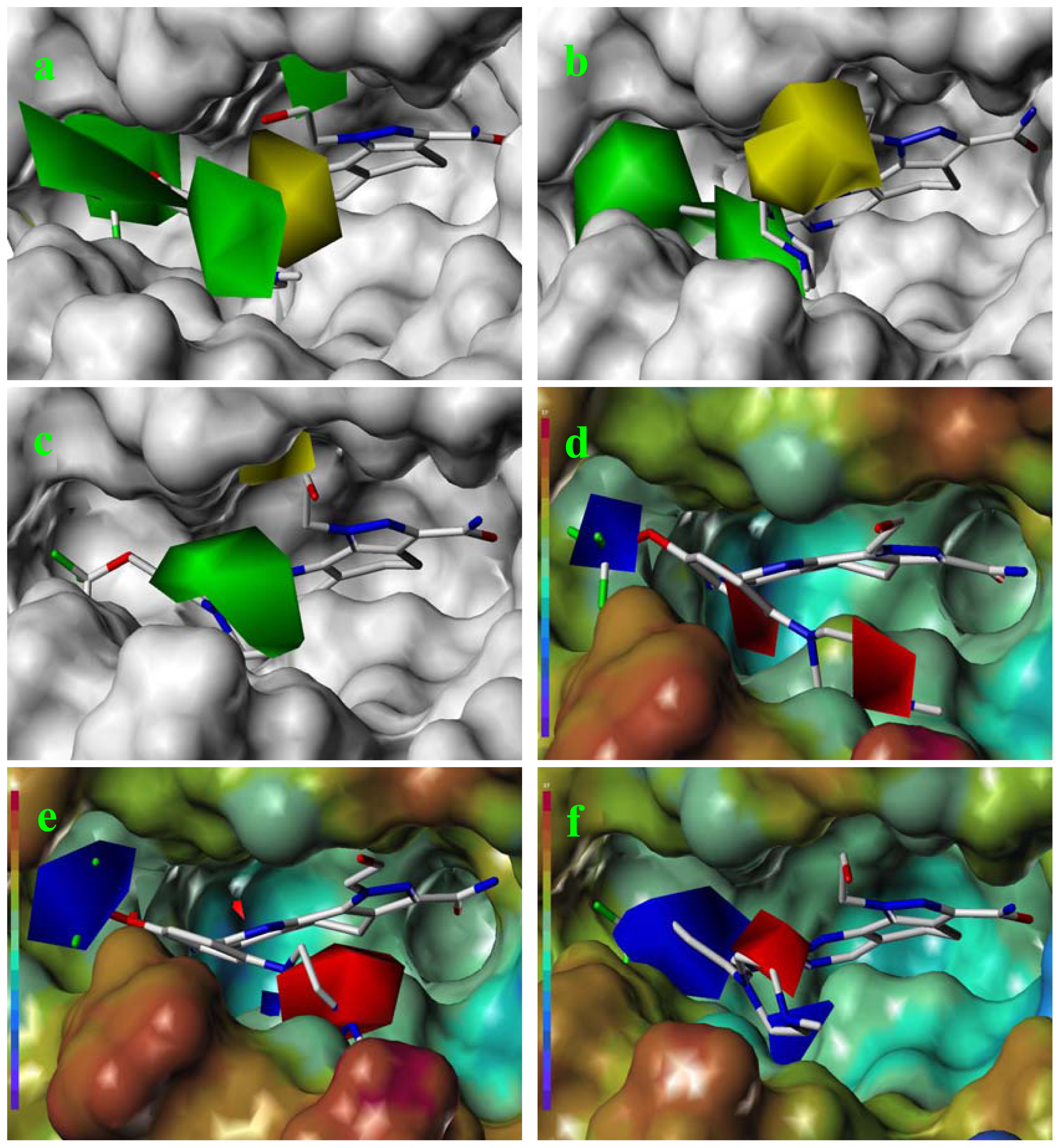
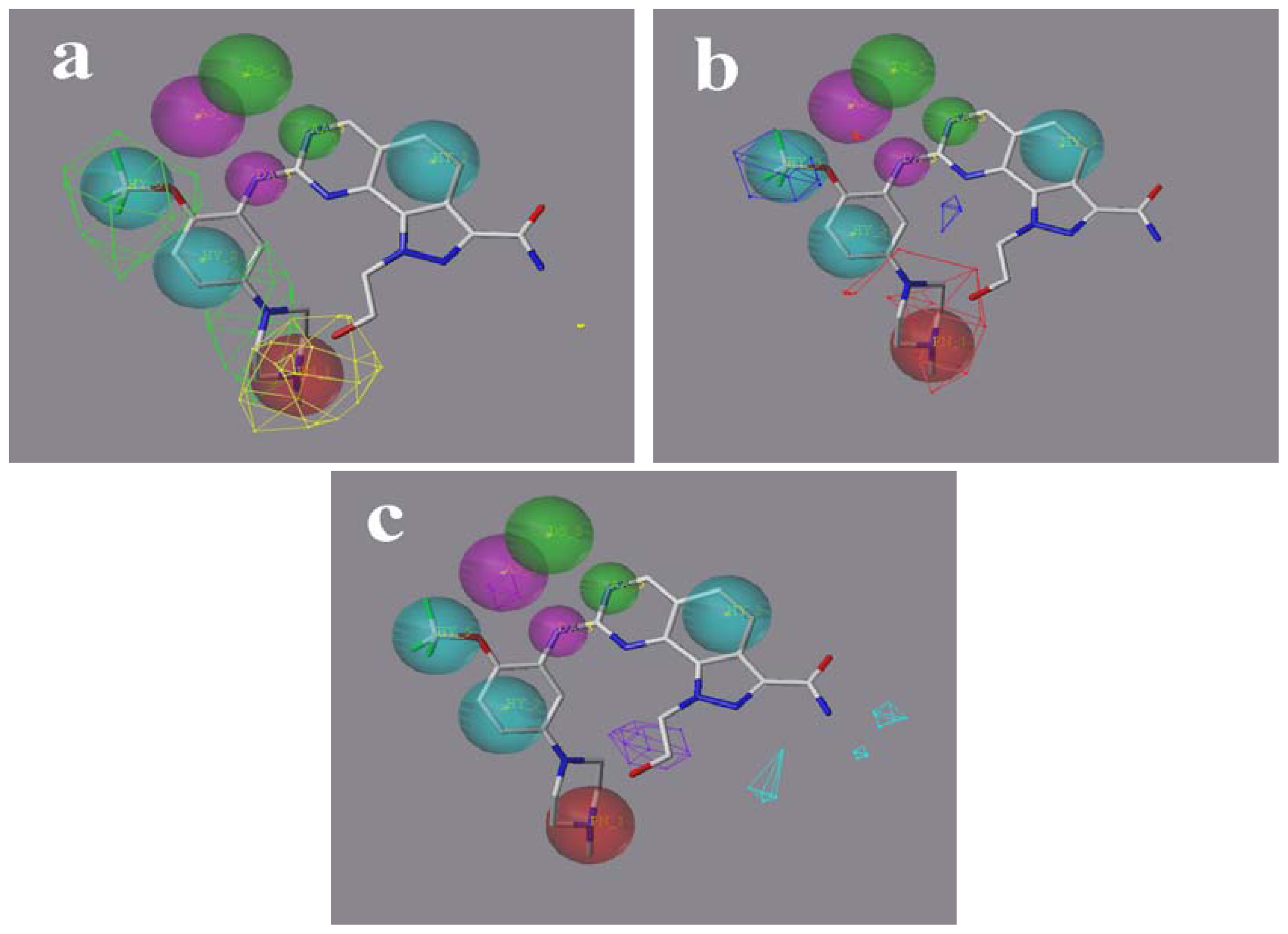
2.5. Comparison of Pharmacophore Model and CoMSIA Model
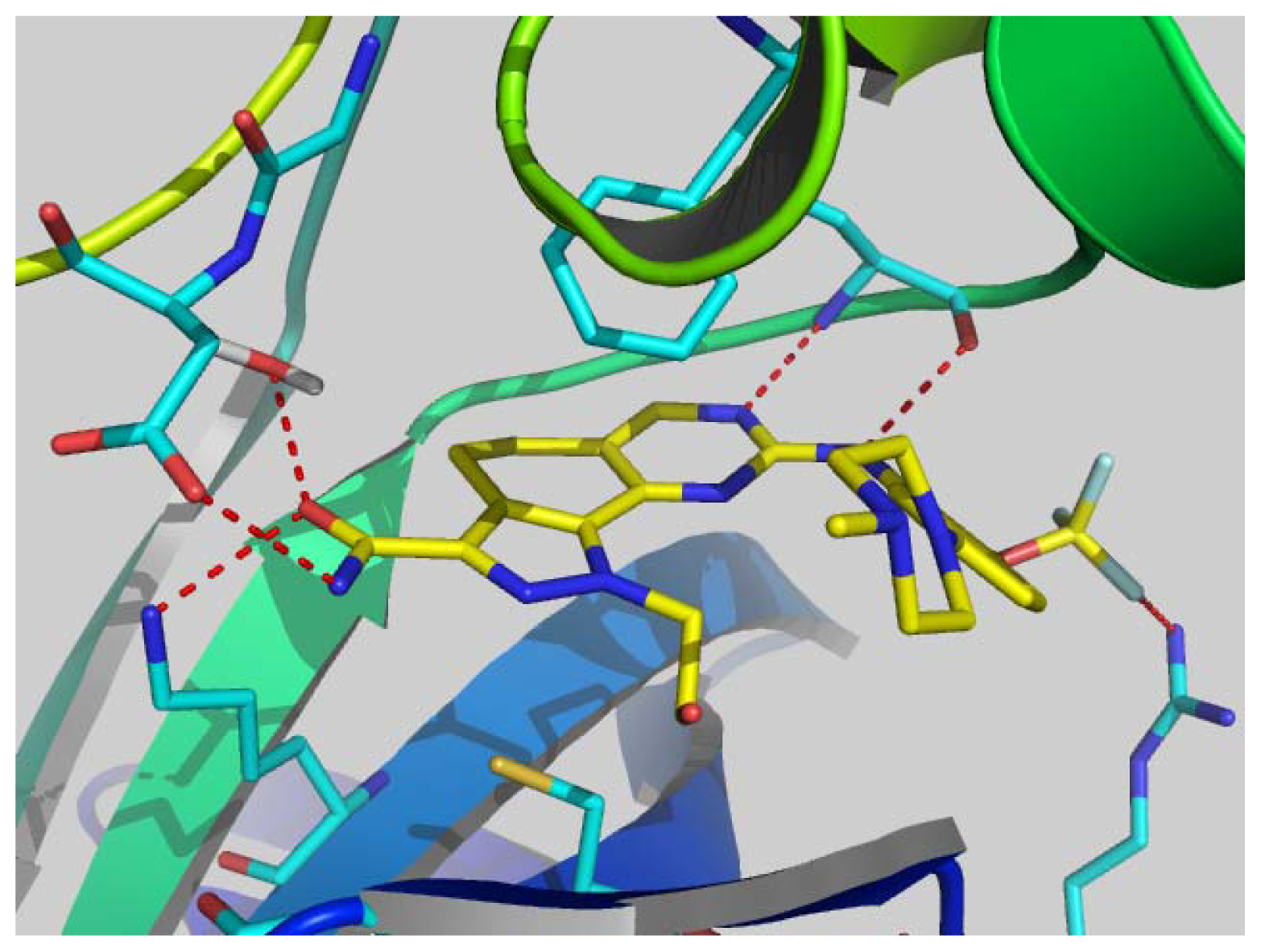
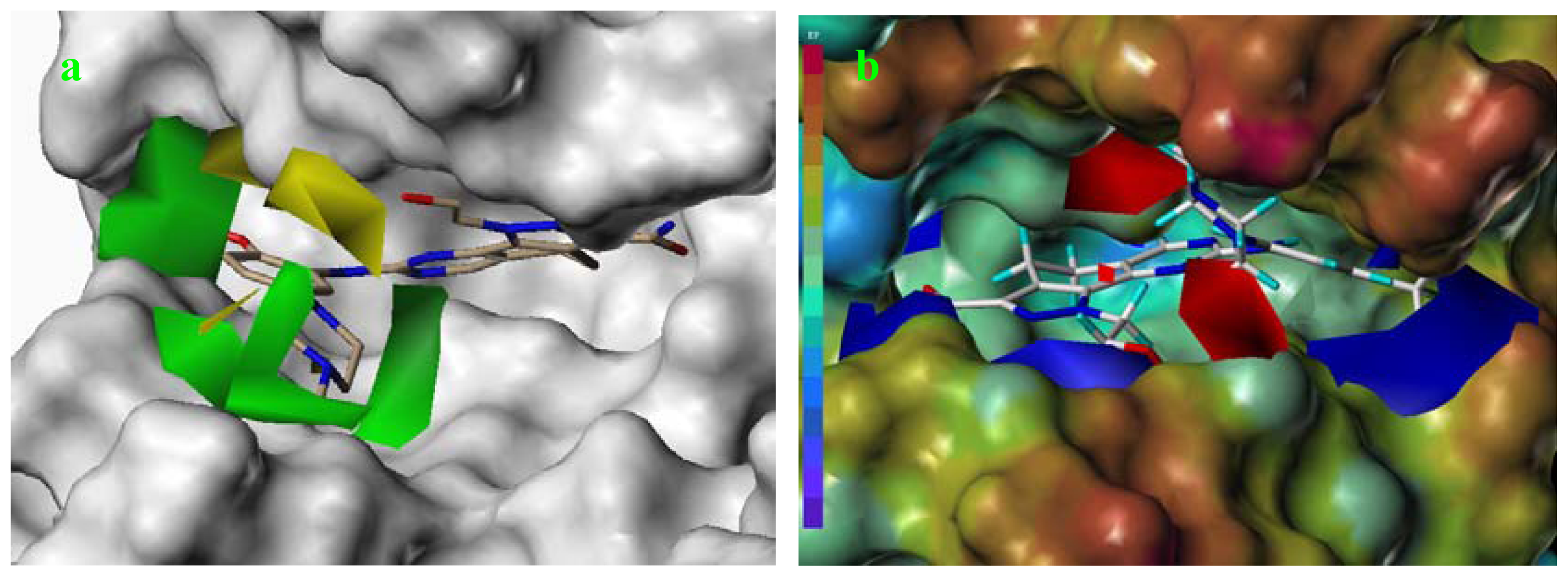
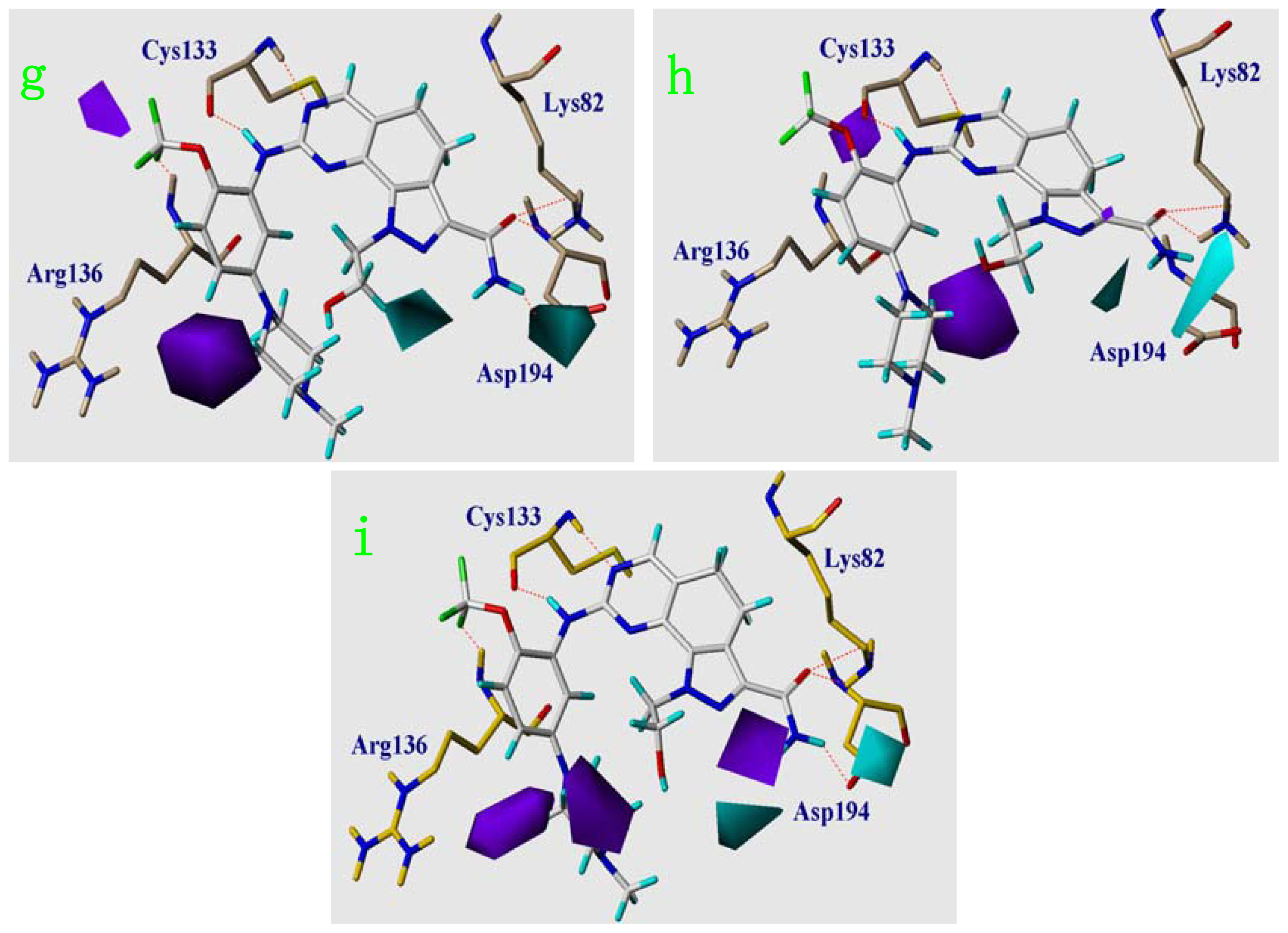
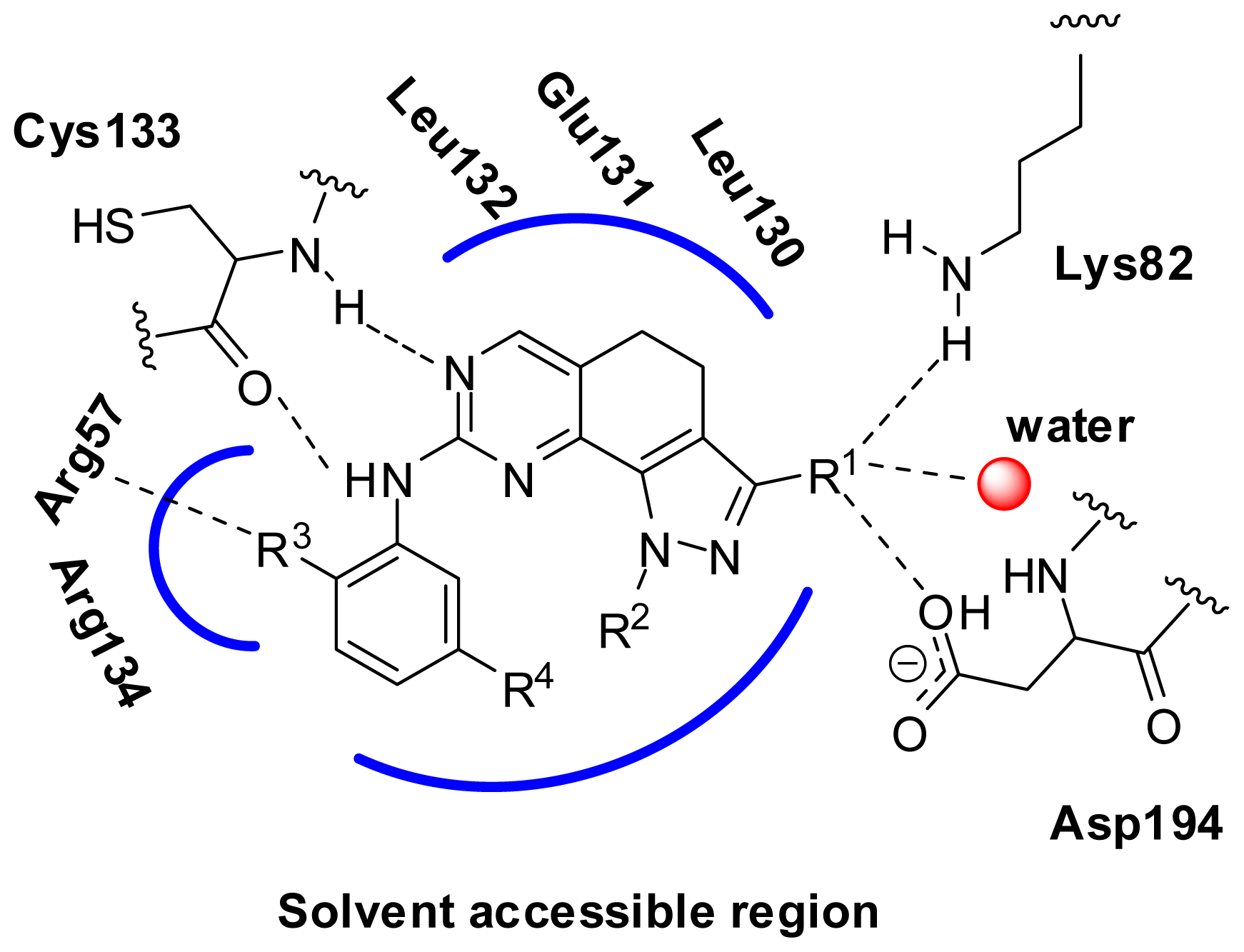
2.6. Structural Insights from 3D-QSAR and Pharmacophore Studies
3. Materials and Methods
3.1. Dataset
3.2. Conformational Alignment
3.2.1. Common Substructure Based Alignment
3.2.2. Molecular Docking Based Alignment
3.2.3. Pharmacophore Based Alignment
3.3. CoMFA and CoMSIA Methodology
3.4. Partial Least Squared (PLS) Analyses and Validation
4. Conclusions
Acknowledgments
References
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of Polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar]
- de Carcer, G.; Escobar, B.; Higuero, A.M.; Garcia, L.; Anson, A.; Perez, G.; Mollejo, M.; Manning, G.; Melendez, B.; Abad-Rodriguez, J.; et al. Plk5, a polo box domain-only protein with specific roles in neuron differentiation and glioblastoma suppression. Mol. Cell. Biol 2011, 31, 1225–1239. [Google Scholar]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like kinase in cancer therapy. Clin. Cancer Res 2010, 16, 384–389. [Google Scholar]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar]
- Strebhardt, K. Multifaceted polo-like kinases: Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov 2010, 9, 643–660. [Google Scholar]
- Cogswell, J.P.; Brown, C.E.; Bisi, J.E.; Neill, S.D. Dominant-negative polo-like kinase 1 induces mitotic catastrophe independent of cdc25C function. Cell Growth Differ 2000, 11, 615–623. [Google Scholar]
- Petronczki, M.; Glotzer, M.; Kraut, N.; Peters, J.M. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev. Cell 2007, 12, 713–725. [Google Scholar]
- Kotani, S.; Tugendreich, S.; Fujii, M.; Jorgensen, P.M.; Watanabe, N.; Hoog, C.; Hieter, P.; Todokoro, K. PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell 1998, 1, 371–380. [Google Scholar]
- Qian, Y.W.; Erikson, E.; Li, C.; Maller, J.L. Activated polo-like kinase Plx1 is required at multiple points during mitosis in Xenopus laevis. Mol. Cell. Biol 1998, 18, 4262–4271. [Google Scholar]
- Smits, V.A.; Klompmaker, R.; Arnaud, L.; Rijksen, G.; Nigg, E.A.; Medema, R.H. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat. Cell Biol 2000, 2, 672–676. [Google Scholar]
- Dai, W.; Wang, Q.; Traganos, F. Polo-like kinases and centrosome regulation. Oncogene 2002, 21, 6195–6200. [Google Scholar]
- Burkard, M.E.; Randall, C.L.; Larochelle, S.; Zhang, C.; Shokat, K.M.; Fisher, R.P.; Jallepalli, P.V. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4383–4388. [Google Scholar]
- Moshe, Y.; Boulaire, J.; Pagano, M.; Hershko, A. Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc. Natl. Acad. Sci. USA 2004, 101, 7937–7942. [Google Scholar]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol 2007, 17, 316–322. [Google Scholar]
- Elez, R.; Piiper, A.; Kronenberger, B.; Kock, M.; Brendel, M.; Hermann, E.; Pliquett, U.; Neumann, E.; Zeuzem, S. Tumor regression by combination antisense therapy against Plk1 and Bcl-2. Oncogene 2003, 22, 69–80. [Google Scholar]
- Spankuch-Schmitt, B.; Wolf, G.; Solbach, C.; Loibl, S.; Knecht, R.; Stegmuller, M.; von Minckwitz, G.; Kaufmann, M.; Strebhardt, K. Downregulation of human polo-like kinase activity by antisense oligonucleotides induces growth inhibition in cancer cells. Oncogene 2002, 21, 3162–3171. [Google Scholar]
- Takai, N.; Hamanaka, R.; Yoshimatsu, J.; Miyakawa, I. Polo-like kinases (Plks) and cancer. Oncogene 2005, 24, 287–291. [Google Scholar]
- Beria, I.; Bossi, R.T.; Brasca, M.G.; Caruso, M.; Ceccarelli, W.; Fachin, G.; Fasolini, M.; Forte, B.; Fiorentini, F.; Pesenti, E.; et al. NMS-P937, a 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivative as potent and selective Polo-like kinase 1 inhibitor. Bioorg. Med. Chem. Lett 2011, 21, 2969–2974. [Google Scholar]
- McInnes, C.; Wyatt, M.D. PLK1 as an oncology target: Current status and future potential. Drug Discov. Today 2011, 16, 619–625. [Google Scholar]
- Beria, I.; Ballinari, D.; Bertrand, J.A.; Borghi, D.; Bossi, R.T.; Brasca, M.G.; Cappella, P.; Caruso, M.; Ceccarelli, W.; Ciavolella, A.; et al. Identification of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as a new class of orally and selective Polo-like kinase 1 inhibitors. J. Med. Chem 2010, 53, 3532–3551. [Google Scholar]
- Beria, I.; Valsasina, B.; Brasca, M.G.; Ceccarelli, W.; Colombo, M.; Cribioli, S.; Fachin, G.; Ferguson, R.D.; Fiorentini, F.; Gianellini, L.M.; et al. 4,5-Dihydro-1H-pyrazolo[4,3-h]quinazolines as potent and selective Polo-like kinase 1 (PLK1) inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 6489–6494. [Google Scholar]
- Keppner, S.; Proschak, E.; Schneider, G.; Spankuch, B. Identification and validation of a potent type II inhibitor of inactive polo-like kinase 1. ChemMedChem 2009, 4, 1806–1809. [Google Scholar]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc 1988, 110, 5959–5967. [Google Scholar]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem 1994, 37, 4130–4146. [Google Scholar]
- SYBYL, version 6.9, Users’ Manual; Tripos, Inc.: St. Louis, MO, USA, 2002.
- Fernandez, A.; Sanguino, A.; Peng, Z.; Crespo, A.; Ozturk, E.; Zhang, X.; Wang, S.; Bornmann, W.; Lopez-Berestein, G. Rational drug redesign to overcome drug resistance in cancer therapy: Imatinib moving target. Cancer Res 2007, 67, 4028–4033. [Google Scholar]
- Wu, G.; Robertson, D.H.; Brooks, C.L., III; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem 2003, 24, 1549–1562. [Google Scholar]
- Discovery Studio, version 2.5, Help Topics; Accelrys Inc.: San Diego, CA, USA, 2009.
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 1997, 267, 727–748. [Google Scholar]
- GOLD, version 5.0 User Guide; CCDC Software Ltd.: Cambridge, UK, 2010.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [Google Scholar]
- GLIDE, version 4.5 User Manual; Schrödinger, LLC: New York, NY, USA, 2007.
- Leach, A.R.; Gillet, V.J.; Lewis, R.A.; Taylor, R. Three-dimensional pharmacophore methods in drug discovery. J. Med. Chem 2010, 53, 539–558. [Google Scholar]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model 2004, 45, 160–169. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 |
| 1 * | NHMe | Me | H | H |
| 2 * | NHcyclopropyl | Me | H | H |
| 3 | NHcyclopentyl | Me | H | H |
| 4 | NHPh | Me | H | H |
| 5 | NH2 | Me | CF3 | H |
| 6 | NH2 | Ph | H | H |
| 7 | NH2 | i-Pr | H | H |
| 8 | NH2 | 1-methylpiperidine-4-yl | H | H |
| 9 | NH2 | 2-(piperidin-1-yl)-ethyl | H | H |
| 10* | NH2 | Me | H | m-CF3 |
| 11 | NH2 | Me | H | p-CF3 |
| 12 | NH2 | Me | Ac | H |
| 13 | NH2 | Me | H | m-Ac |
| 14 | NH2 | Me | OMe | H |
| 15 | NH2 | Me | H | m-OMe |
| 16 | NH2 | Me | H | p-OMe |
| 17 | NH2 | Me | NO2 | H |
| 18 | NH2 | Me | H | m-NO2 |
| 19 | NH2 | Me | Me | H |
| 20 * | NH2 | Me | NHMe | H |
| 21 | NH2 | Me | i-Pr | H |
| 22 * | NH2 | Me | COOMe | H |
| 23 * | NH2 | Me | CONH2 | H |
| 24 | NH2 | Me | SO2NH2 | H |
| 25 | NH2 | Me | Ph | H |
| 26 * | NH2 | Me | OPh | H |
| 27 | NH2 | Me | benzyl | H |
| 28 * | NH2 | Me | NHPh | H |
| 29 | NH2 | Me | benzoyl | H |
| 30 * | NH2 | Me | SPh | H |
| 31 | NH2 | Me | Ac | 3′-(4-methyl-piperazin-1-yl) |
| 32 * | NH2 | Me | Ac | 4′-(4-methyl-piperazin-1-yl) |
| 33 | NH2 | Me | Ac | 5′-(4-methyl-piperazin-1-yl) |
| 34 | NH2 | Me | OMe | 4′-(4-methyl-piperazin-1-yl) |
| 35 * | NH2 | Me | OMe | 5′-(4-methyl-piperazin-1-yl) |
| 36 | OEt | Me | H | H |
| 37 | NH2 | Me | NH2 | H |
| 38 | NH2 | Me | NHAc | H |
| 39 | NH2 | Me | OCF3 | H |
| 40 | NH2 | Me | OCF3 |  |
| 41 * | NH2 | Me | OCF3 |  |
| 42 | NH2 | Me | OCF3 |  |
| 43 | NH2 | Me | OCF3 |  |
| 44 | NH2 | Me | OCF3 |  |
| 45 | NH2 | Me | OCF3 |  |
| 46 * | NH2 | Trityl | OCF3 |  |
| 47 | NH2 | H | OCF3 |  |
| 48 * | NH2 | 2-Fluoro-ethyl | OCF3 |  |
| 49 | NH2 | Ethyl | OCF3 |  |
| 50 * | NH2 | 2-Methoxy-ethyl | OCF3 |  |
| 51 | NH2 | 2-Chloro-ethyl | OCF3 |  |
| 52 | NH2 | Vinyl | OCF3 |  |
| 53 * | NH2 | Me | OCF3 |  |
| 54 | NH2 | Me | OCF3 | 5′-NH2 |
| 55 | NH2 | Me | OCF3 |  |
| 56 * | NH2 | Me | OCF3 |  |
| 57 | NH2 | Me | OCF3 |  |
| 58 | NH2 | Me | OCF3 |  |
| 59 | NH2 | Me | OCF3 |  |
| 60 | NH2 | Me | OCF3 |  |
| 61 | NH2 | Me | OCF3 |  |
| 62 * | NH2 | Me | OCF3 |  |
| 63 | NH2 | Me | OCF3 |  |
| 64 * | NH2 | Me | OCF3 |  |
| 65 | NH2 | Me | OCF3 |  |
| 66 * | NH2 | Me | OCF3 |  |
| 67 | NH2 | Me | OCF3 |  |
| 68 | NH2 | Me | OCF3 |  |
| 69 | NH2 | −(CH2)3–N–(CH3)2 | OCF3 |  |
| 70 * | NH2 |  | OCF3 |  |
| 71 | NH2 | − (CH2)2–NH2 | OCF3 |  |
| 72 | NH2 | − (CH2)3–NH2 | OCF3 |  |
| 73 | NH2 | − (CH2)2–OH | OCF3 |  |
| Alignment Method | CoMFA Model | CoMSIA Model | ||||
|---|---|---|---|---|---|---|
| GD a | PH b | CS c | GD | PH | CS | |
| No. | 1 | 2 | 3 | 4 | 5 | 6 |
| q2 | 0.283 | 0.628 | 0.578 | 0.574 | 0.532 | 0.588 |
| r2 | 0.42 | 0.941 | 0.867 | 0.97 | 0.859 | 0.834 |
| SEE d | 0.818 | 0.268 | 0.404 | 0.198 | 0.411 | 0.447 |
| F e | 36.929 | 192.635 | 78.313 | 207.094 | 99.622 | 81.965 |
| ONC f | 1 | 4 | 4 | 7 | 3 | 3 |
| Field analysis | ||||||
| Steric | 0.663 | 0.619 | 0.698 | 0.177 | 0.224 | 0.263 |
| Electrostatic | 0.337 | 0.381 | 0.618 | 0.52 | 0.407 | 0.459 |
| Hydrophobic | - | - | - | - | - | - |
| H-bond donor | - | - | - | 0.303 | 0.369 | 0.277 |
| H-bond acceptor | - | - | - | - | - | - |
| Test set | ||||||
| r2pred g | 0.405 | 0.785 | 0.752 | 0.605 | 0.695 | 0.749 |
| Compound | pIC50 | Model 1 | Model 2 | Model 3 | |||
|---|---|---|---|---|---|---|---|
| Prediction | Residue | Prediction | Residue | Prediction | Residue | ||
| 1 * | 5.375 | 5.848 | −0.473 | 5.227 | 0.148 | 5.380 | −0.005 |
| 2 * | 5.000 | 5.934 | −0.934 | 5.406 | −0.406 | 5.063 | −0.063 |
| 3 | 5.000 | 5.824 | −0.824 | 5.228 | −0.228 | 4.778 | 0.222 |
| 4 | 5.000 | 5.724 | −0.724 | 4.795 | 0.205 | 5.298 | −0.298 |
| 5 | 6.365 | 6.544 | −0.180 | 6.382 | −0.018 | 6.282 | 0.082 |
| 6 | 5.000 | 6.101 | −1.101 | 5.591 | −0.591 | 5.653 | −0.653 |
| 7 | 6.367 | 6.074 | 0.293 | 5.935 | 0.432 | 5.765 | 0.602 |
| 8 | 5.000 | 6.394 | −1.394 | 5.096 | −0.096 | 4.798 | 0.202 |
| 9 | 5.000 | 6.543 | −1.543 | 4.966 | 0.034 | 5.060 | −0.060 |
| 10 * | 7.292 | 6.249 | 1.043 | 7.358 | −0.066 | 6.423 | 0.869 |
| 11 | 6.060 | 5.547 | 0.513 | 6.153 | −0.093 | 6.165 | −0.106 |
| 12 | 6.461 | 6.536 | −0.075 | 6.401 | 0.060 | 6.286 | 0.175 |
| 13 | 7.000 | 5.914 | 1.086 | 7.105 | −0.105 | 6.575 | 0.425 |
| 14 | 7.377 | 6.378 | 0.999 | 7.359 | 0.018 | 7.120 | 0.257 |
| 15 | 6.870 | 6.068 | 0.802 | 6.798 | 0.072 | 6.649 | 0.221 |
| 16 | 6.592 | 5.970 | 0.622 | 6.402 | 0.190 | 6.886 | −0.294 |
| 17 | 6.312 | 6.354 | −0.042 | 6.395 | −0.083 | 6.125 | 0.187 |
| 18 | 5.000 | 5.892 | −0.892 | 5.074 | −0.074 | 6.189 | −1.189 |
| 19 | 7.824 | 6.040 | 1.784 | 7.420 | 0.404 | 6.858 | 0.966 |
| 20 * | 6.959 | 6.260 | 0.699 | 6.240 | 0.719 | 7.193 | −0.234 |
| 21 | 6.438 | 6.216 | 0.222 | 6.485 | −0.047 | 6.749 | −0.311 |
| 22 * | 5.952 | 6.490 | −0.538 | 6.844 | −0.892 | 6.745 | −0.793 |
| 23 * | 5.683 | 6.250 | −0.567 | 5.481 | 0.202 | 6.781 | −1.098 |
| 24 | 5.428 | 6.150 | −0.722 | 5.564 | −0.136 | 6.110 | −0.682 |
| 25 | 5.806 | 6.316 | −0.511 | 6.198 | −0.393 | 6.044 | −0.238 |
| 26 * | 6.556 | 6.624 | −0.068 | 6.279 | 0.277 | 6.216 | 0.340 |
| 27 | 6.026 | 6.216 | −0.191 | 6.005 | 0.021 | 6.035 | −0.010 |
| 28 * | 6.023 | 6.609 | −0.586 | 6.150 | −0.127 | 6.212 | −0.189 |
| 29 | 5.706 | 6.380 | −0.674 | 5.686 | 0.020 | 5.724 | −0.018 |
| 30 * | 5.692 | 6.772 | −1.080 | 6.168 | −0.476 | 6.094 | −0.402 |
| 31 | 5.688 | 5.964 | −0.276 | 5.904 | −0.216 | 5.913 | −0.225 |
| 32 * | 6.334 | 6.768 | −0.435 | 5.746 | 0.587 | 7.694 | −1.361 |
| 33 | 6.963 | 7.711 | −0.748 | 7.152 | −0.189 | 6.698 | 0.265 |
| 34 | 7.398 | 6.327 | 1.071 | 7.573 | −0.175 | 7.330 | 0.068 |
| 35 * | 8.155 | 7.441 | 0.714 | 8.503 | −0.348 | 8.226 | −0.071 |
| 36 | 5.000 | 5.874 | −0.874 | 5.070 | −0.070 | 5.195 | −0.195 |
| 37 | 6.824 | 6.153 | 0.671 | 6.920 | −0.096 | 6.796 | 0.028 |
| 38 | 5.598 | 5.973 | −0.375 | 5.145 | 0.453 | 5.588 | 0.010 |
| 39 | 6.932 | 6.387 | 0.545 | 6.681 | 0.251 | 6.716 | 0.216 |
| 40 | 6.016 | 7.373 | −1.357 | 6.026 | −0.010 | 5.755 | 0.261 |
| 41 * | 6.910 | 7.384 | −0.474 | 6.320 | 0.590 | 7.188 | −0.278 |
| 42 | 8.097 | 7.501 | 0.596 | 8.130 | −0.033 | 8.653 | −0.556 |
| 43 | 7.114 | 7.318 | −0.204 | 7.163 | −0.050 | 7.056 | 0.058 |
| 44 | 8.699 | 7.428 | 1.271 | 8.567 | 0.132 | 8.275 | 0.424 |
| 45 | 7.456 | 7.522 | −0.066 | 7.163 | 0.293 | 7.905 | −0.449 |
| 46 * | 5.000 | 7.379 | −2.379 | 5.528 | −0.528 | 4.123 | 0.877 |
| 47 | 8.398 | 7.156 | 1.242 | 8.361 | 0.037 | 8.363 | 0.035 |
| 48 * | 8.398 | 7.276 | 1.122 | 7.868 | 0.530 | 8.064 | 0.334 |
| 49 | 8.222 | 7.609 | 0.613 | 7.959 | 0.263 | 8.069 | 0.153 |
| 50 * | 7.824 | 7.300 | 0.524 | 7.527 | 0.297 | 7.675 | 0.149 |
| 51 | 8.155 | 7.316 | 0.839 | 8.152 | 0.003 | 7.918 | 0.237 |
| 52 | 8.222 | 7.518 | 0.704 | 8.339 | −0.117 | 8.073 | 0.149 |
| 53 * | 8.523 | 7.555 | 0.968 | 8.221 | 0.302 | 8.579 | −0.056 |
| 54 | 7.222 | 6.432 | 0.790 | 7.500 | −0.278 | 7.182 | 0.040 |
| 55 | 7.602 | 7.416 | 0.186 | 7.967 | −0.365 | 7.685 | −0.083 |
| 56 * | 7.482 | 7.453 | 0.028 | 6.750 | 0.732 | 8.656 | −1.175 |
| 57 | 6.699 | 7.505 | −0.806 | 7.303 | −0.604 | 6.568 | 0.131 |
| 58 | 8.523 | 7.497 | 1.026 | 8.082 | 0.441 | 8.499 | 0.024 |
| 59 | 8.222 | 7.333 | 0.889 | 8.070 | 0.152 | 8.018 | 0.204 |
| 60 | 6.813 | 7.516 | −0.704 | 6.580 | 0.233 | 7.415 | −0.603 |
| 61 | 6.697 | 7.502 | −0.805 | 6.439 | 0.258 | 7.358 | −0.661 |
| 62 * | 7.377 | 7.417 | −0.040 | 7.465 | −0.088 | 7.735 | −0.358 |
| 63 | 7.222 | 7.129 | 0.093 | 7.568 | −0.346 | 6.608 | 0.614 |
| 64 * | 6.975 | 7.354 | −0.379 | 6.220 | 0.755 | 7.077 | −0.102 |
| 65 | 6.827 | 7.660 | −0.833 | 6.591 | 0.236 | 7.034 | −0.207 |
| 66 * | 7.081 | 7.358 | −0.277 | 6.546 | 0.535 | 6.968 | 0.113 |
| 67 | 7.357 | 7.555 | −0.199 | 7.285 | 0.071 | 6.833 | 0.523 |
| 68 | 6.650 | 7.634 | −0.984 | 6.181 | 0.469 | 6.117 | 0.533 |
| 69 | 6.554 | 7.291 | −0.737 | 6.763 | −0.209 | 6.665 | −0.111 |
| 70 * | 7.367 | 7.521 | −0.155 | 6.587 | 0.780 | 7.756 | −0.390 |
| 71 | 7.482 | 7.562 | −0.081 | 8.013 | −0.532 | 7.765 | −0.284 |
| 72 | 7.284 | 7.647 | −0.363 | 7.332 | −0.048 | 7.703 | −0.419 |
| 73 | 8.699 | 7.541 | 1.158 | 8.292 | 0.407 | 8.235 | 0.464 |
| Compound | pIC50 | Model 4 | Model 5 | Model 6 | |||
|---|---|---|---|---|---|---|---|
| Prediction | Residue | Prediction | Residue | Prediction | Residue | ||
| 1 * | 5.375 | 5.171 | 0.204 | 5.208 | 0.167 | 4.833 | 0.542 |
| 2 * | 5.000 | 5.524 | −0.524 | 5.168 | −0.168 | 4.781 | 0.219 |
| 3 | 5.000 | 5.037 | −0.037 | 5.159 | −0.159 | 4.620 | 0.380 |
| 4 | 5.000 | 5.099 | −0.099 | 5.024 | −0.024 | 5.314 | −0.314 |
| 5 | 6.365 | 6.248 | 0.116 | 6.307 | 0.057 | 6.225 | 0.140 |
| 6 | 5.000 | 4.967 | 0.033 | 5.446 | −0.446 | 5.907 | −0.907 |
| 7 | 6.367 | 6.230 | 0.137 | 5.931 | 0.436 | 5.911 | 0.456 |
| 8 | 5.000 | 5.102 | −0.102 | 5.189 | −0.189 | 4.950 | 0.050 |
| 9 | 5.000 | 5.239 | −0.239 | 5.014 | −0.014 | 5.194 | −0.194 |
| 10 * | 7.292 | 6.092 | 1.200 | 6.508 | 0.784 | 6.233 | 1.059 |
| 11 | 6.060 | 6.063 | −0.003 | 5.964 | 0.095 | 6.148 | −0.088 |
| 12 | 6.461 | 6.453 | 0.008 | 6.401 | 0.060 | 6.052 | 0.409 |
| 13 | 7.000 | 7.012 | −0.012 | 6.302 | 0.698 | 6.818 | 0.182 |
| 14 | 7.377 | 7.070 | 0.307 | 7.380 | −0.003 | 6.911 | 0.466 |
| 15 | 6.870 | 7.040 | −0.170 | 6.452 | 0.418 | 6.517 | 0.353 |
| 16 | 6.592 | 6.660 | −0.068 | 6.576 | 0.016 | 6.926 | −0.334 |
| 17 | 6.312 | 6.395 | −0.083 | 6.653 | −0.341 | 6.169 | 0.143 |
| 18 | 5.000 | 5.062 | −0.062 | 5.449 | −0.449 | 5.846 | −0.846 |
| 19 | 7.824 | 7.011 | 0.813 | 6.951 | 0.873 | 6.576 | 1.248 |
| 20 * | 6.959 | 6.528 | 0.431 | 5.916 | 1.043 | 6.666 | 0.293 |
| 21 | 6.438 | 6.699 | −0.261 | 6.742 | −0.304 | 6.645 | −0.207 |
| 22 * | 5.952 | 6.461 | −0.509 | 6.993 | −1.041 | 6.700 | −0.748 |
| 23 * | 5.683 | 5.490 | 0.193 | 5.590 | 0.093 | 6.630 | −0.947 |
| 24 | 5.428 | 5.327 | 0.101 | 5.574 | −0.146 | 5.521 | −0.093 |
| 25 | 5.806 | 5.896 | −0.090 | 6.583 | −0.778 | 6.265 | −0.459 |
| 26 * | 6.556 | 5.894 | 0.662 | 6.111 | 0.445 | 6.740 | −0.184 |
| 27 | 6.026 | 5.814 | 0.212 | 6.347 | −0.322 | 6.295 | −0.270 |
| 28 * | 6.023 | 5.744 | 0.279 | 6.477 | −0.454 | 6.749 | −0.726 |
| 29 | 5.706 | 5.708 | −0.002 | 5.322 | 0.384 | 6.148 | −0.442 |
| 30 * | 5.692 | 6.086 | −0.394 | 6.111 | −0.419 | 6.328 | −0.636 |
| 31 | 5.688 | 5.601 | 0.087 | 6.117 | −0.429 | 6.026 | −0.338 |
| 32 * | 6.334 | 8.044 | −1.711 | 6.289 | 0.045 | 7.253 | −0.920 |
| 33 | 6.963 | 6.937 | 0.026 | 6.742 | 0.221 | 6.561 | 0.402 |
| 34 | 7.398 | 7.349 | 0.049 | 7.880 | −0.482 | 7.183 | 0.215 |
| 35 * | 8.155 | 7.191 | 0.964 | 7.979 | 0.176 | 8.207 | −0.052 |
| 36 | 5.000 | 5.047 | −0.047 | 4.871 | 0.129 | 5.260 | −0.260 |
| 37 | 6.824 | 6.803 | 0.021 | 6.603 | 0.221 | 7.167 | −0.343 |
| 38 | 5.598 | 5.651 | −0.053 | 5.731 | −0.133 | 5.382 | 0.216 |
| 39 | 6.932 | 7.623 | −0.691 | 6.973 | −0.041 | 6.569 | 0.363 |
| 40 | 6.016 | 6.033 | −0.017 | 6.427 | −0.411 | 5.770 | 0.246 |
| 41 * | 6.910 | 6.012 | 0.898 | 6.753 | 0.157 | 7.180 | −0.270 |
| 42 | 8.097 | 8.147 | −0.050 | 8.069 | 0.028 | 8.308 | −0.211 |
| 43 | 7.114 | 7.115 | −0.002 | 6.944 | 0.170 | 6.619 | 0.495 |
| 44 | 8.699 | 8.774 | −0.075 | 8.472 | 0.227 | 8.358 | 0.341 |
| 45 | 7.456 | 7.337 | 0.119 | 6.944 | 0.512 | 7.701 | −0.245 |
| 46 * | 5.000 | 5.778 | −0.778 | 5.855 | −0.855 | 4.419 | 0.581 |
| 47 | 8.398 | 8.364 | 0.034 | 8.644 | −0.246 | 8.514 | −0.116 |
| 48 * | 8.398 | 8.319 | 0.079 | 8.168 | 0.230 | 7.910 | 0.488 |
| 49 | 8.222 | 8.041 | 0.181 | 7.951 | 0.271 | 7.944 | 0.278 |
| 50 * | 7.824 | 7.912 | −0.088 | 7.834 | −0.010 | 7.619 | 0.205 |
| 51 | 8.155 | 8.241 | −0.086 | 7.889 | 0.266 | 7.766 | 0.389 |
| 52 | 8.222 | 8.378 | −0.156 | 8.125 | 0.097 | 7.931 | 0.291 |
| 53 * | 8.523 | 8.002 | 0.521 | 8.126 | 0.397 | 8.239 | 0.284 |
| 54 | 7.222 | 7.369 | −0.147 | 7.428 | −0.206 | 7.069 | 0.153 |
| 55 | 7.602 | 7.615 | −0.013 | 8.039 | −0.437 | 7.530 | 0.072 |
| 56 * | 7.482 | 7.503 | −0.022 | 7.064 | 0.418 | 8.246 | −0.765 |
| 57 | 6.699 | 6.573 | 0.126 | 6.999 | −0.300 | 6.315 | 0.384 |
| 58 | 8.523 | 8.441 | 0.082 | 7.866 | 0.657 | 8.501 | 0.022 |
| 59 | 8.222 | 8.102 | 0.120 | 8.128 | 0.094 | 7.886 | 0.336 |
| 60 | 6.813 | 6.696 | 0.117 | 6.928 | −0.116 | 7.765 | −0.953 |
| 61 | 6.697 | 6.888 | −0.191 | 6.960 | −0.263 | 7.513 | −0.816 |
| 62 * | 7.377 | 7.339 | 0.038 | 7.754 | −0.377 | 7.886 | −0.509 |
| 63 | 7.222 | 7.253 | −0.031 | 7.863 | −0.641 | 6.816 | 0.406 |
| 64 * | 6.975 | 6.454 | 0.521 | 5.790 | 1.185 | 7.407 | −0.432 |
| 65 | 6.827 | 6.790 | 0.037 | 6.234 | 0.593 | 7.473 | −0.646 |
| 66 * | 7.081 | 6.289 | 0.792 | 6.296 | 0.785 | 7.145 | −0.064 |
| 67 | 7.357 | 7.293 | 0.063 | 6.725 | 0.632 | 6.899 | 0.458 |
| 68 | 6.650 | 6.515 | 0.135 | 6.238 | 0.412 | 6.724 | −0.074 |
| 69 | 6.554 | 6.540 | 0.014 | 7.249 | −0.695 | 7.200 | −0.646 |
| 70 * | 7.367 | 6.809 | 0.558 | 6.531 | 0.836 | 7.257 | 0.110 |
| 71 | 7.482 | 7.571 | −0.090 | 7.993 | −0.512 | 7.728 | −0.247 |
| 72 | 7.284 | 7.357 | −0.073 | 7.314 | −0.030 | 7.624 | −0.340 |
| 73 | 8.699 | 8.680 | 0.019 | 7.805 | 0.894 | 8.294 | 0.405 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lu, S.; Liu, H.-C.; Chen, Y.-D.; Yuan, H.-L.; Sun, S.-L.; Gao, Y.-P.; Yang, P.; Zhang, L.; Lu, T. Combined Pharmacophore Modeling, Docking, and 3D-QSAR Studies of PLK1 Inhibitors. Int. J. Mol. Sci. 2011, 12, 8713-8739. https://doi.org/10.3390/ijms12128713
Lu S, Liu H-C, Chen Y-D, Yuan H-L, Sun S-L, Gao Y-P, Yang P, Zhang L, Lu T. Combined Pharmacophore Modeling, Docking, and 3D-QSAR Studies of PLK1 Inhibitors. International Journal of Molecular Sciences. 2011; 12(12):8713-8739. https://doi.org/10.3390/ijms12128713
Chicago/Turabian StyleLu, Shuai, Hai-Chun Liu, Ya-Dong Chen, Hao-Liang Yuan, Shan-Liang Sun, Yi-Ping Gao, Pei Yang, Liang Zhang, and Tao Lu. 2011. "Combined Pharmacophore Modeling, Docking, and 3D-QSAR Studies of PLK1 Inhibitors" International Journal of Molecular Sciences 12, no. 12: 8713-8739. https://doi.org/10.3390/ijms12128713