Identification of (−)(E)-N-[2(S)-Hydroxy-2-(4-hydroxyphenyl) ethyl]ferulamide, a Natural Product Isolated from Croton Pullei: Theoretical and Experimental Analysis

Abstract
:1. Introduction
2. Results and Discussion
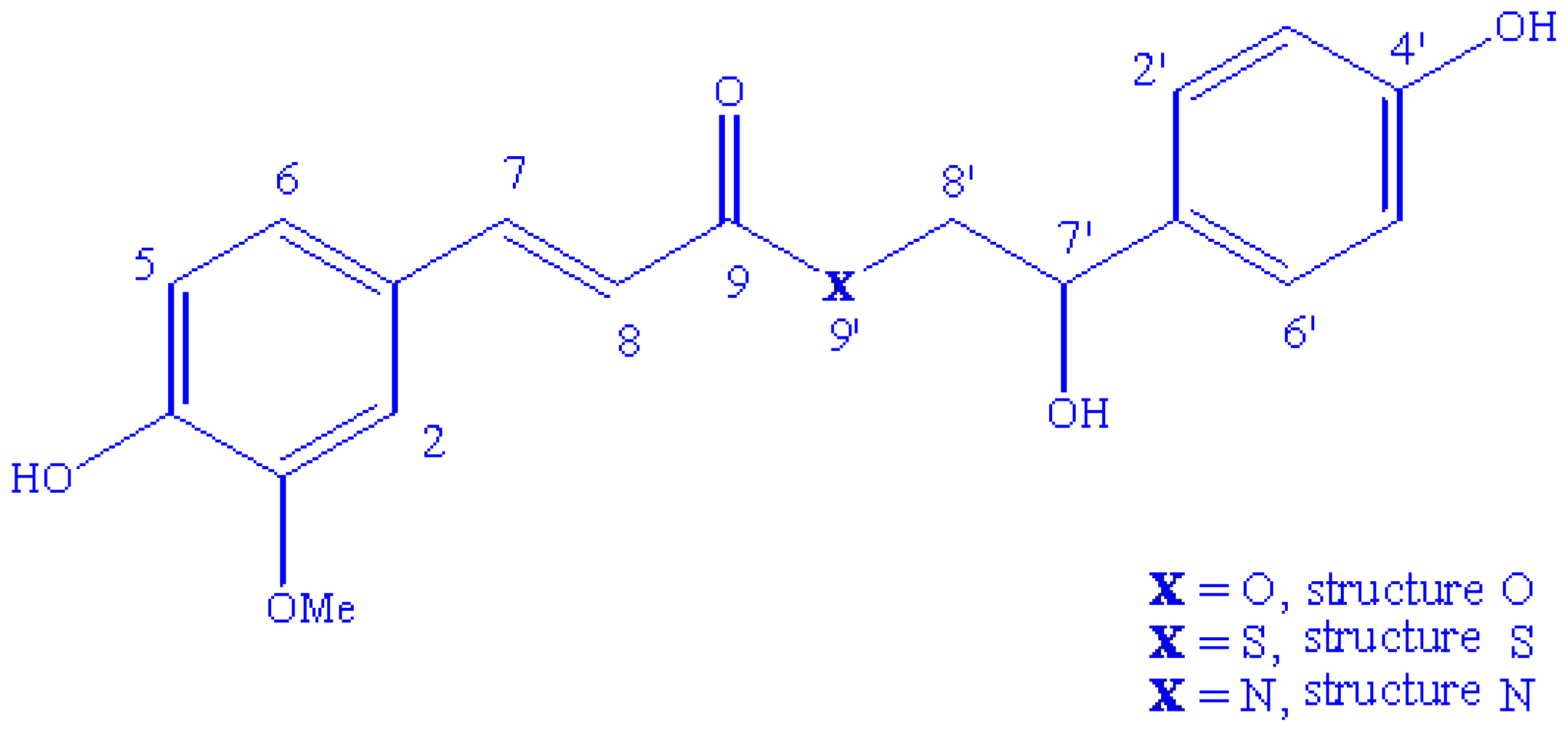
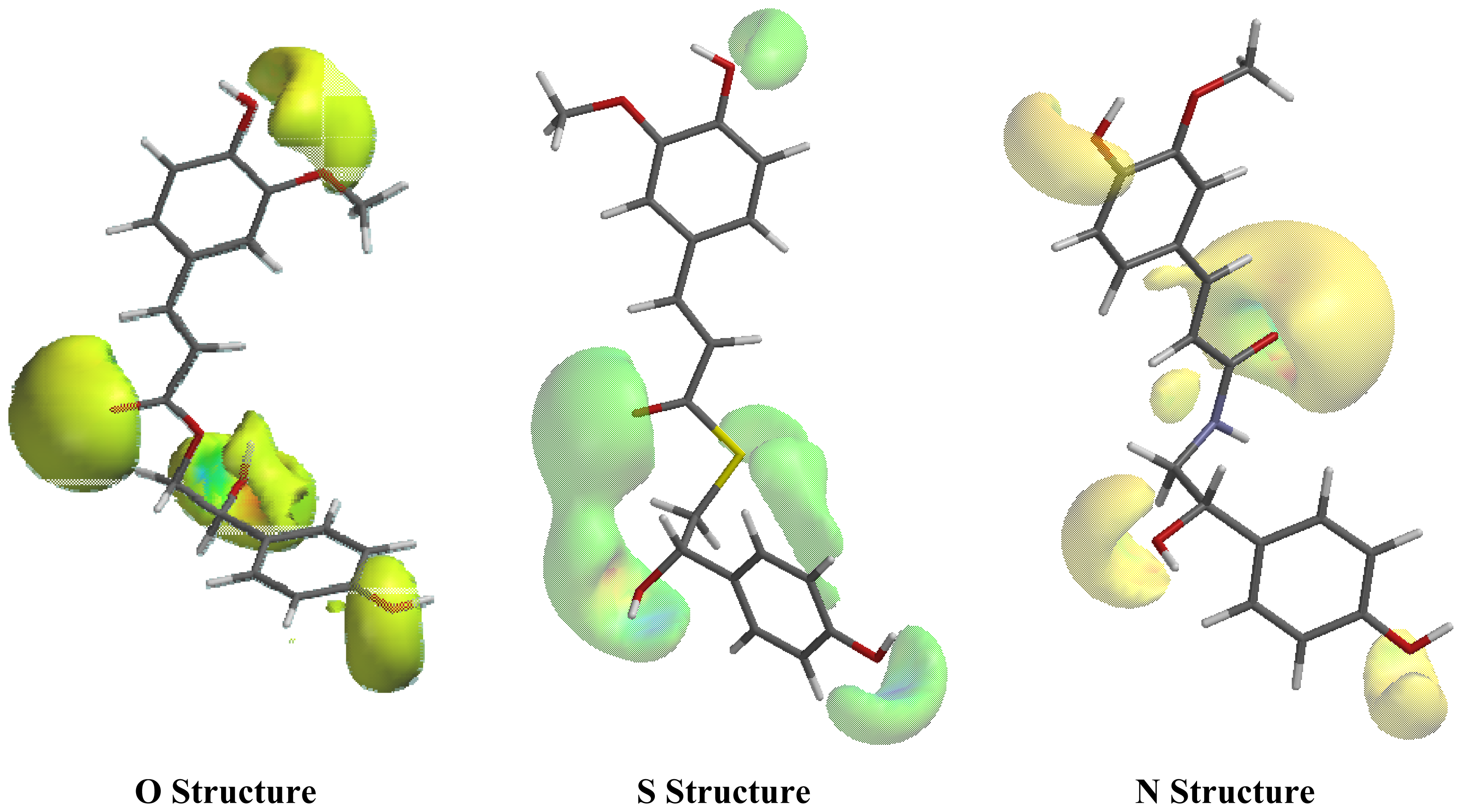
2.1. Conformational Analysis and Geometric Data
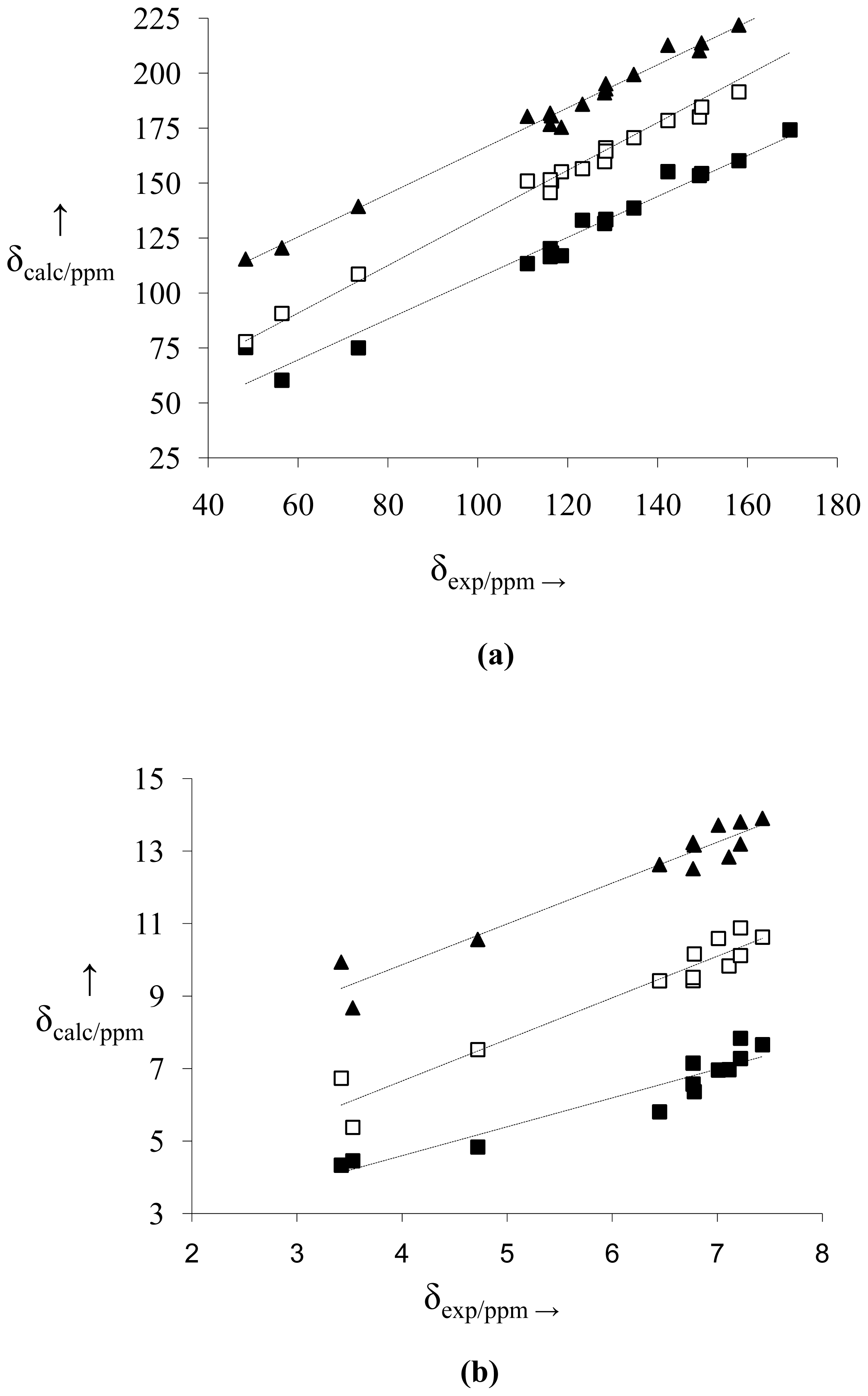
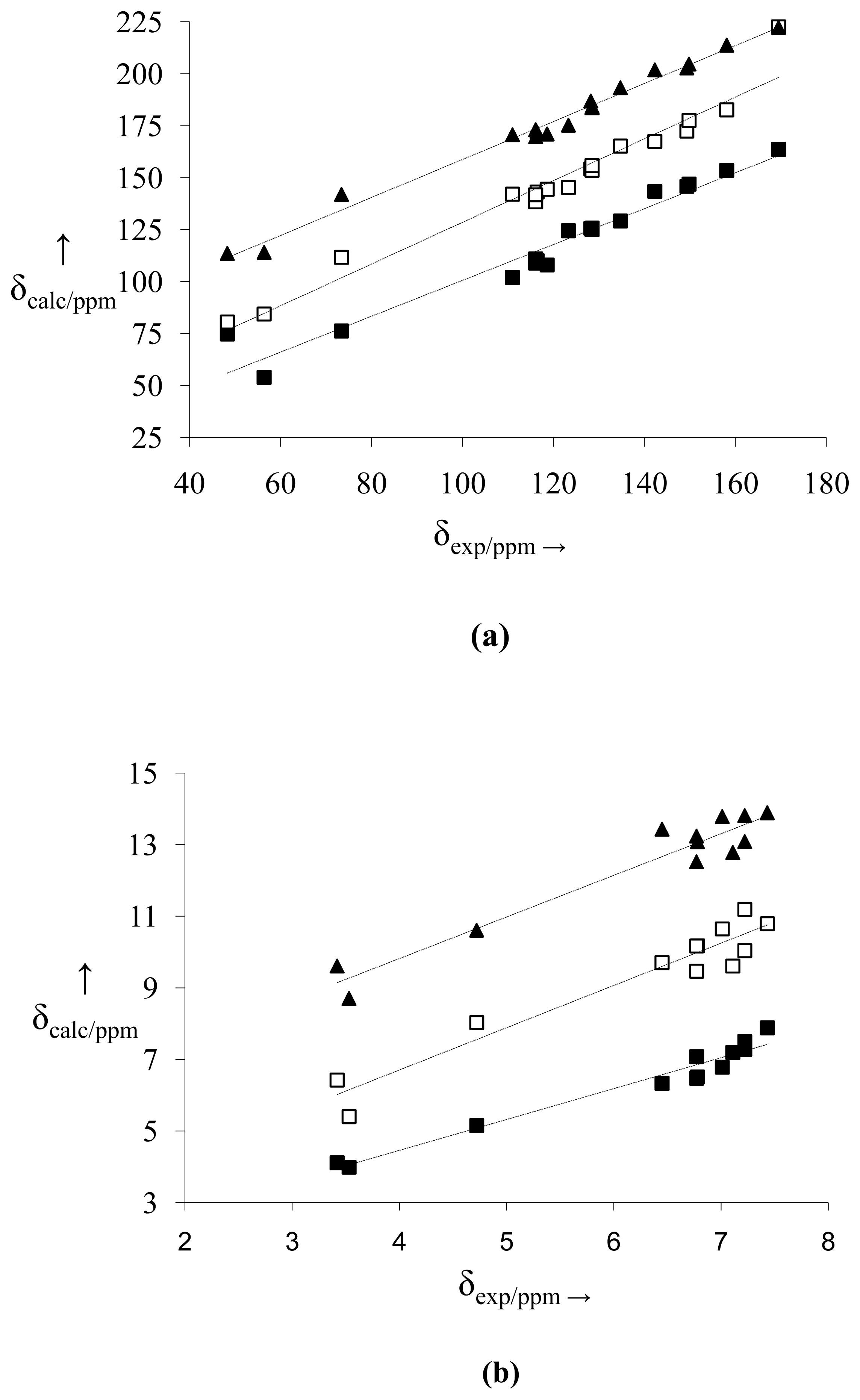
2.2. NMR Spectra and Statistical Analysis
2.3. Infrared Spectrum
2.4. Polarimetry
3. Experimental Section
3.1. Collection and Extraction
3.2. Computational Method
3.3. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Sarangi, P.K.; Nanda, S.; Sahoo, H. Enhancing the rate of ferulic acid bioconversion using different carbon sources. J. Brew. Distill 2011, 2, 1–4. [Google Scholar]
- Anselmi, C.; Bernardi, F.; Centini, M.; Gaggelli, E.; Gaggelli, N.; Valensin, D.; Valensin, G.M. Interaction of ferulic acid derivatives with human erythrocytes monitored by pulse field gradient NMR diffusion and NMR relaxation studies. Chem. Phys. Lipids 2005, 134, 109–117. [Google Scholar]
- Anselmi, C.; Centini, M.; Maggiore, M.; Gaggelli, N.; Andreassi, M.; Buonocore, A.; Beretta, G.; Facino, R.M. Non-covalent inclusion of ferulic acid with α-cyclodextrin improves photo-stability and delivery: NMR and modeling studies. J. Pharm. Biomed. Anal 2008, 46, 645–652. [Google Scholar]
- Graf, E. Antioxidant potential of ferulic acid. Free Radic. Biol. Med 1992, 13, 435–448. [Google Scholar]
- Rakotondramanana, D.L.A.; Delomenède, M.; Baltas, M.; Duran, H.; Bedos-Belval, F.; Rasoanaivo, P.; Negre-Salvayre, A.; Gornitzka, H. Synthesis of ferulic ester dimers, functionalisation and biological evaluation as potential antiatherogenic and antiplasmodial agents. Bioorg. Med. Chem 2007, 15, 6018–6026. [Google Scholar]
- Chemistry of Natural Products. Available online: http://quiprona.wordpress.com/2009/06/17/analises-por-rmn-alguns-cuidados accessed on 13 April 2011.
- Carvalho, I.; Pupo, M.T.; Borges, Á.D.L.; Bernardes, L.S.C. Introdução a modelagem molecular de fármacos no curso experimental de química farmacêutica. Quim. Nova 2003, 26, 428–438. [Google Scholar]
- Tikhele, S.H.; Pissurlenkar, R.R.S.; Srivastava, S.; Saran, A.; Coutinho, E.C. Mapping interactions of gastric inhibitory polypeptide with GIPR N-terminus using NMR and molecular dynamics simulations. J. Pept. Sci 2010, 16, 383–391. [Google Scholar]
- Silva, J.R.A.; Lameira, J.; Santana, P.P.B.; Silva, A.; Schneider, M.P.C.; Alves, C.N. Homology modeling and molecular dynamics simulation of an alpha methyl coenzyme M reductase from Methanogenic archea. Int. J. Quantum Chem 2010, 110, 2067–2075. [Google Scholar]
- Plaza, A.; Piacente, S.; Perrone, A.; Hamed, A.; Pizza, C.; Bifulco, G. Stemmosides C and D, two novel unusual pregnane glycosides from Solenostemma argel: Structural elucidation and configurational study by a combined NMR-quantum mechanical strategy. Tetrahedron 2004, 52, 12201–12209. [Google Scholar]
- Wang, F.D.; Wang, T.; Wu, A.A.; Ding, L.; Wang, H.Q. X-ray and DFT study of glaucocalyxin A compound with cytotoxic activity. Chin. J. Chem. Phys 2009, 22, 275–284. [Google Scholar]
- Wang, T.; Tang, F.; Zhang, Y.; Chen, Z. A natural diterpenoid kamebacetal A with anti-tumor activity: Theoretical and experimental study. J. Mol. Struct 2010, 975, 317–322. [Google Scholar]
- Azizoglu, A.; Ozer, Z.; Kilic, T. An experimental and theoretical study on siderol isolated from Sideritis species. Collect. Czech. Chem. Commun. 2011, 76, 95–114. [Google Scholar]
- Moreira, R.Y.O.; Brasil, D.S.B.; Alves, C.N.; Guilhon, G.M.S.P.; Santos, L.S.; Arruda, M.S.P.; Muller, A.H.; Barbosa, P.S.; Abreu, A.S.; Silva, E.O.; et al. Crystal structure and theoretical calculations of julocrotine, a natural product with antileishmanial activity. Int. J. Quantum Chem 2008, 108, 513–520. [Google Scholar]
- Barbosa, P.S.; Abreu, A.; Batista, E.F.; Guilhon, G.M.S.P.; Muller, A.H.; Arruda, M.S.P.; Santos, L.S.; Arruda, A.C.; Secco, R.S. Glutarimide alkaloids and terpenoids from Croton pullei var glabrior Lanj. Biochem. Syst. Ecol 2007, 35, 887–890. [Google Scholar]
- Abreu, A.S.; Barbosa, P.S.; Müller, A.H.; Guilhon, G.M.S.P. Constituintes químicos do caule e das cascas do caule de Croton pullei var. glabrior (Euphorbiaceae). Rev. Virtual Iniciaç. Acad. UFPA 2001, 1, 1–9. [Google Scholar]
- Lee, H.; Oh, S.; Kwon, O.; Ahn, K.; Lee, J.; Kim, J.; Min, B.; Joung, H. Isolation of coumarins and ferulate from the roots of Angelica purpuraefolia and the antitumor activity of khellactone. Phytother. Res 2007, 21, 406–409. [Google Scholar]
- Dellagreca, M.; Previtera, L.; Purcaro, R.; Zarrelli, A. Cinnamic acid amides and lignanamides from Aptenia cordifolia. Tetrahedron 2006, 62, 2877–2882. [Google Scholar]
- Lee, D.G.; Park, Y.; Kim, M.; Jung, H.; Seu, Y.; Hahm, K.; Woo, E. Anti-fungal effects of phenolic amides isolated from the root bark of Lycium chinense. Biotechnol. Lett 2004, 26, 1125–1130. [Google Scholar]
- Nesterenko, V.; Putt, K.S.; Hergenrother, J. Identification from a combinatorial library of a small molecule that selectively induces apoptosis in cancer Cells. J. Am. Chem. Soc 2003, 125, 14672–14673. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the colle-salvetti correlation-energy formula into a funtional of the electron density. Phys. Rev 1998, 37, 785–789. [Google Scholar]
- Prachayasittikul, S.; Suphapong, S.; Worachartcheewan, A.; Lawung, R.; Ruchirawat, S.; Prachayasittikul, V. Bioactive Metabolites from Spilanthes acmella Murr. Molecules 2009, 14, 850–867. [Google Scholar]
- HyperChem, version 7.5; Hypercube, Inc: Gainesville, FL, USA, 2002.
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem 1989, 10, 209–220. [Google Scholar]
- Smith, S.G.; Goodman, J.M. Assigning the stereochemistry of paris of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem 2009, 74, 4597–4607. [Google Scholar]
- Franks, W.T.; Zhou, D.H.; Wylie, B.J.; Money, B.G.; Graesser, D.T.; Frericks, H.L.; Sahota, G.; Rienstra, C.M. Magic-angle spinning solid-state nmr spectroscopy of the β1 immunoglobulin binding domain of protein G (GB1): 15N and 13C chemical shift assignments and conformational analysis. J. Am. Chem. Soc 2005, 127, 12291–12305. [Google Scholar]
- Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford, CT, USA, 2004.
- Brasil, D.S.B.; Alves, C.N.; Guilhon, G.M.S.P.; Muller, A.H.; Secco, R.S.; Gabriel, R.; Llusar, P. Crystal structure and theoretical study of IR and 1H and 13C NMR spectra of cordatin, a natural product with antiulcerogenic activity. Int. J. Quantum Chem. 2008, 108, 2564–2575. [Google Scholar]
- Brasil, D.S.B.; Muller, A.H.; Guilhon, G.M.S.P.; Alves, C.N.; Peris, G.; Llusard, R.; Molinerd, V. Isolation, X-ray Crystal structure and theoretical calculations of the new compound 8-epicordatin and identification of others terpenes and steroids from the bark and leaves of Croton palanostigma Klotzsch. J. Braz. Chem. Soc 2010, 21, 731–739. [Google Scholar]
- Spartan software, version 8; Wavefunction, Inc: Irvine, CA, USA, 2008.
- MINITAB, MINITAB Release 14; MINITAB: State College, PA, USA, 2003.
- Cimino, P.; Gomez-Paloma, L.; Duca, D.; Riccio, R.; Bifulco, G. Comparison of different theory models and basis sets in the calculation of 13C NMR chemical shifts of natural products. Magn. Reson. Chem 2004, 42, S26–S33. [Google Scholar]
- Gaudio, A.C.; Zandonade, E. Proposição, validação e análise dos modelos que correlacionam estrutura química e atividade biológica. Quim. Nova 2001, 24, 658–671. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3PW91/DGDZVP | B3LYP/6-31+G(d,p) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | Exp. | Structure O | Structure S | Structure N | Structure O | Structure S | Structure N | ||||||
| Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | ||
| 1 | 128.2 | 131.6 | 3.4 | 129.8 | 1.6 | 131.0 | 2.8 | 125.7 | 2.5 | 124.2 | 4.0 | 126.8 | 1.4 |
| 2 | 111.0 | 113.4 | 2.3 | 121.0 | 10.0 | 120.3 | 9.3 | 101.9 | 9.0 | 112.1 | 1.1 | 110.7 | 0.3 |
| 3 | 149.3 | 153.5 | 4.2 | 150.2 | 0.8 | 150.2 | 0.9 | 145.9 | 3.4 | 142.5 | 6.9 | 142.8 | 6.4 |
| 4 | 149.8 | 154.4 | 4.6 | 154.7 | 4.9 | 153.7 | 3.9 | 146.9 | 2.9 | 147.5 | 2.3 | 144.6 | 5.2 |
| 5 | 116.4 | 118.3 | 1.9 | 120.9 | 4.5 | 120.6 | 4.2 | 110.2 | 6.2 | 113.2 | 3.2 | 111.9 | 4.5 |
| 6 | 123.3 | 133.2 | 9.9 | 126.6 | 3.3 | 125.9 | 2.6 | 124.4 | 1.1 | 115.3 | 8.0 | 115.2 | 8.1 |
| 7 | 142.3 | 155.2 | 12.9 | 148.5 | 6.2 | 152.7 | 10.4 | 143.5 | 1.2 | 137.5 | 4.8 | 141.8 | 0.5 |
| 8 | 118.6 | 117.0 | 1.6 | 125.3 | 6.7 | 115.4 | 3.2 | 108.0 | 10.6 | 114.4 | 4.2 | 110.9 | 7.6 |
| 9 | 169.5 | 174.2 | 4.7 | 200.1 | 30.6 | 171.4 | 1.9 | 163.6 | 5.9 | 192.5 | 23.0 | 162.3 | 7.2 |
| 1′ | 134.7 | 138.7 | 4.0 | 140.7 | 6.0 | 139.4 | 4.7 | 129.2 | 5.5 | 135.2 | 0.5 | 133.3 | 1.4 |
| 2′ | 128.5 | 133.6 | 5.1 | 136.0 | 7.5 | 132.9 | 4.4 | 125.8 | 2.7 | 123.6 | 4.9 | 124.1 | 4.4 |
| 3′ | 116.1 | 120.3 | 4.2 | 115.7 | 0.4 | 121.8 | 5.7 | 110.9 | 5.1 | 108.4 | 7.7 | 113.0 | 3.1 |
| 4′ | 158.1 | 160.3 | 2.3 | 161.6 | 3.4 | 161.9 | 3.8 | 153.4 | 4.7 | 152.6 | 5.5 | 153.7 | 4.4 |
| 5′ | 116.1 | 116.6 | 0.5 | 121.6 | 5.5 | 116.7 | 0.6 | 108.9 | 7.2 | 111.8 | 4.3 | 109.7 | 6.4 |
| 6′ | 128.5 | 133.4 | 4.9 | 134.6 | 6.1 | 135.2 | 6.7 | 125.0 | 3.5 | 125.8 | 2.8 | 123.6 | 4.9 |
| 7′ | 73.4 | 75.1 | 1.7 | 78.6 | 5.2 | 79.4 | 5.9 | 76.2 | 2.8 | 81.7 | 8.3 | 81.9 | 8.6 |
| 8′ | 48.3 | 75.3 | 26.9 | 47.9 | 0.3 | 55.5 | 7.2 | 74.8 | 26.5 | 50.6 | 2.3 | 53.5 | 5.2 |
| OMe | 56.4 | 60.7 | 3.9 | 60.7 | 4.3 | 60.5 | 4.1 | 53.9 | 2.51 | 54.4 | 2.0 | 54.0 | 2.4 |
| B3PW91/DGDZVP | B3LYP/6-31+G(d,p) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Position | Exp. | Structure O | Structure S | Structure N | Structure O | Structure S | Structure N | ||||||
| Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | Calc. | RS | ||
| 2 | 7.11 | 6.97 | 0.14 | 6.83 | 0.28 | 6.83 | 0.28 | 7.19 | 0.08 | 6.61 | 0.50 | 6.78 | 0.33 |
| 5 | 6.78 | 6.37 | 0.41 | 7.16 | 0.38 | 7.17 | 0.39 | 6.51 | 0.27 | 7.17 | 0.39 | 7.08 | 0.30 |
| 6 | 7.01 | 6.96 | 0.05 | 7.59 | 0.58 | 7.71 | 0.70 | 6.78 | 0.23 | 7.65 | 0.64 | 7.79 | 0.78 |
| 7 | 7.43 | 7.66 | 0.23 | 7.63 | 0.20 | 7.90 | 0.47 | 7.88 | 0.45 | 7.79 | 0.36 | 7.89 | 0.46 |
| 8 | 6.45 | 5.81 | 0.64 | 6.42 | 0.03 | 6.62 | 0.17 | 6.33 | 0.12 | 6.71 | 0.26 | 7.43 | 0.98 |
| 2′ | 7.22 | 7.28 | 0.06 | 7.12 | 0.10 | 7.80 | 0.58 | 7.28 | 0.06 | 7.04 | 0.18 | 7.81 | 0.59 |
| 3′ | 6.77 | 7.15 | 0.38 | 6.43 | 0.34 | 7.23 | 0.46 | 7.08 | 0.31 | 6.47 | 0.30 | 7.24 | 0.47 |
| 5′ | 6.77 | 6.57 | 0.20 | 7.21 | 0.44 | 6.51 | 0.26 | 6.47 | 0.30 | 7.17 | 0.40 | 6.52 | 0.25 |
| 6′ | 7.22 | 7.84 | 0.62 | 7.88 | 0.66 | 7.19 | 0.03 | 7.51 | 0.29 | 8.20 | 0.98 | 7.09 | 0.13 |
| 7′ | 4.72 | 4.84 | 0.12 | 4.52 | 0.20 | 4.56 | 0.16 | 5.15 | 0.43 | 5.03 | 0.31 | 4.61 | 0.11 |
| 8′ a | 3.53 | 4.46 | 0.93 | 2.38 | 1.15 | 2.67 | 0.86 | 3.99 | 0.46 | 2.40 | 1.13 | 2.70 | 0.83 |
| 8′ b | 3.42 | 4.33 | 0.91 | 3.73 | 0.31 | 3.94 | 0.52 | 4.11 | 0.69 | 3.43 | 0.01 | 3.61 | 0.19 |
| Position | 13C | 1H | ||||
|---|---|---|---|---|---|---|
| Exp. [22] | Calc. | RS | Exp. [22] | Calc. | RS | |
| 1 | 127.8 | 126.1 | 1.6 | - | - | |
| 2 | 111.6 | 113.5 | 1.8 | 7.2 | 6.7 | 0.5 |
| 3 | 151.5 | 145.1 | 6.4 | - | - | |
| 4 | 149.9 | 145.7 | 4.2 | - | - | |
| 5 | 116.5 | 111.4 | 5.1 | 6.8 | 6.6 | 0.2 |
| 6 | 123.9 | 113.6 | 10.4 | 7.5 | 7.5 | 0.4 |
| C-α | 115.9 | 107.7 | 8.2 | 6.3 | 6.5 | 0.2 |
| C-β | 146.9 | 147.2 | 0.3 | 7.6 | 7.8 | 0.2 |
| C=O | 171.2 | 162.7 | 8.5 | - | - | |
| OMe-3 | 56.5 | 53.9 | 2.5 | 3.9 | 3.8 | 0.1 |
| δ | a | b | R2 | MAE | CMAE | s | PRESS | sPRESS | F | Q2 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| B3PW91/DGDZVP | δ(1H) O | 0.80 | 1.41 | 0.90 | 0.39 | 0.40 | 0.4910 | 3.6794 | 0.19 | 86.11 | 84.12% |
| δ(1H) S | 1.16 | −0.98 | 0.94 | 0.39 | 0.31 | 0.4182 | 3.0896 | 0.18 | 122.49 | 86.67% | |
| δ(1H) N | 1.13 | −0.64 | 0.94 | 0.41 | 0.31 | 0.3856 | 2.6661 | 0.16 | 145.83 | 88.50% | |
| δ(13C) O | 0.93 | 13,87 | 0.96 | 5.49 | 4.32 | 6.3579 | 959.879 | 1.94 | 430.88 | 94.69% | |
| δ(13C) S | 1.08 | −4.11 | 0.97 | 5.97 | 3.53 | 5.7831 | 913.886 | 1.89 | 524.12 | 94.94% | |
| δ(13C) N | 0.98 | 6.86 | 0.99 | 4.57 | 2.21 | 3.2676 | 205.189 | 0.89 | 1675.82 | 98.86% | |
| B3LYP/6- 31+G(d,p) | δ(1H) O | 0.86 | 1.00 | 0.96 | 0.31 | 0.25 | 0.2786 | 1.0415 | 0.10 | 222.41 | 94.23% |
| δ(1H) S | 1.18 | −1.02 | 0.92 | 0.46 | 0.35 | 0.5231 | 4.6355 | 0.22 | 118.15 | 86.78% | |
| δ(1H) N | 1.16 | −0.83 | 0.93 | 0.45 | 0.32 | 0.4879 | 3.8216 | 0.19 | 131.42 | 88.65% | |
| δ(13C) O | 0.86 | 14.22 | 0.95 | 5.74 | 5.42 | 6.7773 | 1331.07 | 2.28 | 293.17 | 90.63% | |
| δ(13C) S | 1.00 | −1.92 | 0.95 | 5.32 | 4.70 | 7.4911 | 1304.14 | 2.26 | 324.63 | 93.18% | |
| δ(13C) N | 0.91 | 7.30 | 0.99 | 4.56 | 2.82 | 3.4479 | 260.415 | 1.01 | 1269.99 | 98.30% | |
| δ(1H) FA | 1.02 | −0.12 | 0.95 | 0.26 | 0.24 | 0.3601 | 0.8689 | 0.23 | 71.56 | 91.13% | |
| δ(13C) FA | 0.97 | −0.28 | 0.98 | 4.90 | 3.12 | 4.0927 | 179.844 | 1.68 | 498.65 | 97.88% | |
| Vibrational Mode (cm−1) | Experimental | Theoretical—B3LYP/6-31G(d,p) | Theoretical—B3LYP/6-31+G(d,p) | ||||
|---|---|---|---|---|---|---|---|
| O structure | S structure | N structure | O structure | S structure | N structure | ||
| υ (O-H aliphatic) | 3370 | 3782 | 3808 | 3810 | 3806 | 3633 | 3825 |
| υ (O-H aromatic) | 3370 | 3818 | 3760 | 3763 | 3822 | 3765 | 3612 |
| υ (N-H) | 3370 | - | - | 3612 | - | - | 3612 |
| υ (O-H aromatic) | 3370 | 3822 | 3822 | 3822 | 3828 | 3828 | 3770 |
| υa (CH2) + υs (C-H sp2) | - | 3128 | 3156 | 3127 | 3130 | 3137 | 3114 |
| υs (CH2) + υs (C-H sp3) + υs (C-H sp2) | - | 3077 | 3089 | 3076 | 3081 | 3098 | 3094 |
| υa (CH3) + υ (C-H sp2) | - | 3154 | 3157 | 3155 | 3157 | 3159 | 3157 |
| υ (C-H sp2) + υa (C-H sp2 aromatic) | - | 3207 | 3204 | 3206 | 3207 | 3206 | 3201 |
| υs (CH3) | - | 3016 | 3026 | 3025 | 3017 | 3029 | 3027 |
| υ (C-H sp3) + υs (CH2) | 2930 | 3038 | 3018 | 2985 | 3073 | 3075 | 3064 |
| δa (C-H sp2 aromatic) + υs (C-O) | - | 1675 | 1673 | 1673 | 1664 | 1662 | 1662 |
| υ (C=O) + υ (C=C trans) + δ(C-H) [+δ(N-H)] a | 1680 | 1794 | 1755 | 1758 | 1768 | 1701 | 1727 |
| υs (C=C trans) + υ (C=O) + δ (C-H sp2) or [+ δ(N-H)] a | 1600 | 1691 | 1755 | 1685 | 1679 | 1754 | 1674 |
| δa (CH3) | 1510 | 1505 | 1502 | 1502 | 1509 | 1509 | 1510 |
| δs (C-H sp2 aromatic) + υ (C-O secondary alcohol) | 1480 | 1307 | 1307 | 1309 | 1311 | 1309 | 1308 |
| δs (C-H sp2) + υ (C-O secondary alcohol) | 1390 | 1319 | 1316 | 1319 | 1319 | 1327 | 1318 |
| δs (C-H sp2) + δa (CH3) | 1110 | 1225 | 1220 | 1179 | 1220 | 1228 | 1227 |
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Silva, S.d.O.; Peixoto, R.N.S.; Silva, J.R.A.; Alves, C.N.; Guilhon, G.M.S.P.; Santos, L.S.; Brasil, D.d.S.B. Identification of (−)(E)-N-[2(S)-Hydroxy-2-(4-hydroxyphenyl) ethyl]ferulamide, a Natural Product Isolated from Croton Pullei: Theoretical and Experimental Analysis. Int. J. Mol. Sci. 2011, 12, 9389-9403. https://doi.org/10.3390/ijms12129389
Silva SdO, Peixoto RNS, Silva JRA, Alves CN, Guilhon GMSP, Santos LS, Brasil DdSB. Identification of (−)(E)-N-[2(S)-Hydroxy-2-(4-hydroxyphenyl) ethyl]ferulamide, a Natural Product Isolated from Croton Pullei: Theoretical and Experimental Analysis. International Journal of Molecular Sciences. 2011; 12(12):9389-9403. https://doi.org/10.3390/ijms12129389
Chicago/Turabian StyleSilva, Silvana de O., Rosana N.S. Peixoto, José Rogério A. Silva, Cláudio N. Alves, Giselle M.S.P. Guilhon, Lourivaldo S. Santos, and Davi do S.B. Brasil. 2011. "Identification of (−)(E)-N-[2(S)-Hydroxy-2-(4-hydroxyphenyl) ethyl]ferulamide, a Natural Product Isolated from Croton Pullei: Theoretical and Experimental Analysis" International Journal of Molecular Sciences 12, no. 12: 9389-9403. https://doi.org/10.3390/ijms12129389