Advances in Carcinogenic Metal Toxicity and Potential Molecular Markers

{kind=link}
Abstract
:1. General Features of Carcinogenic Metal Compounds
2. Overall Mechanisms of Metal-Induced Genotoxicity and Carcinogenicity
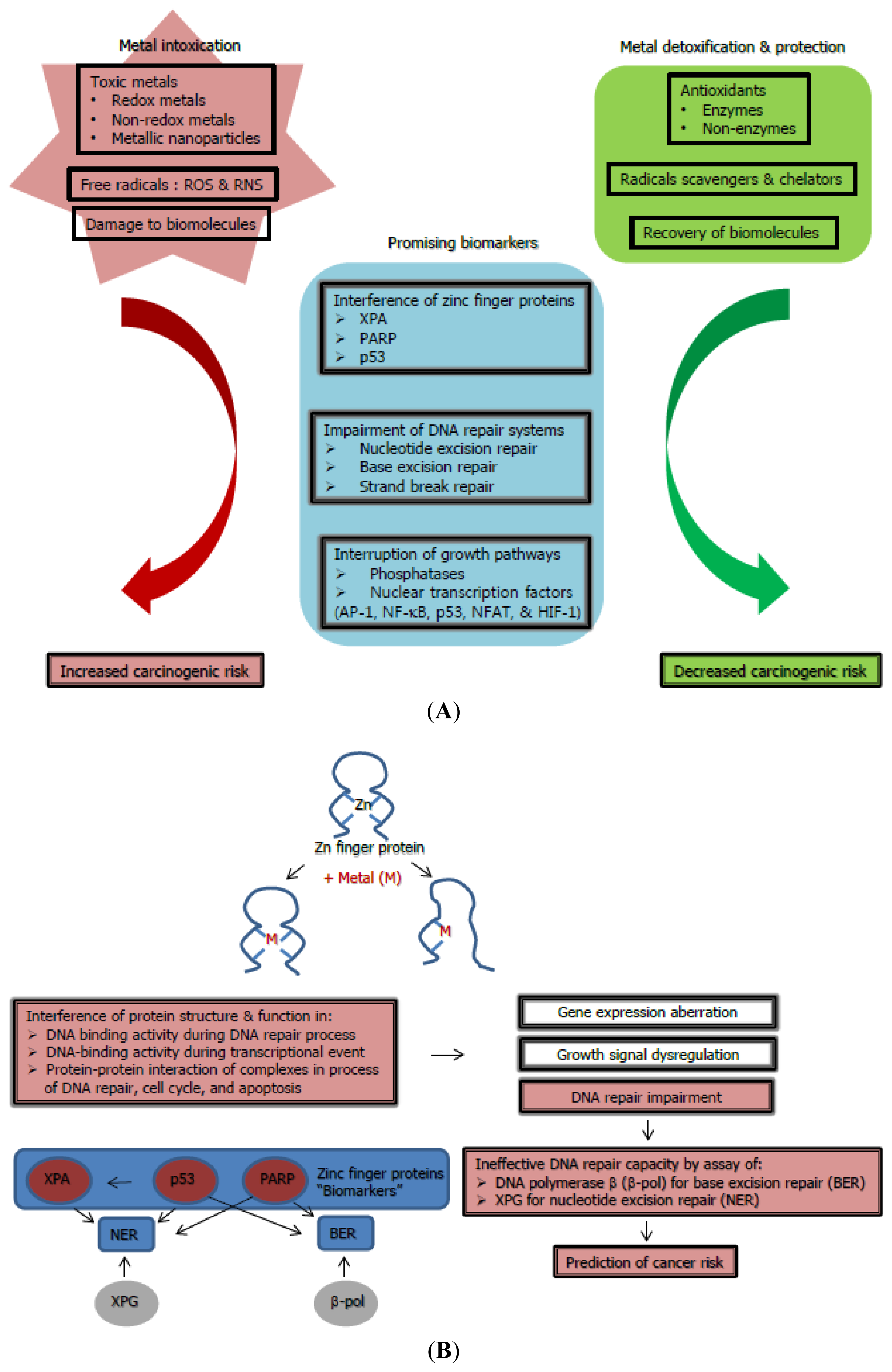
2.1. Induction of Oxidative Stress, a Causative Source for Metal-Toxicity
2.2. Impairment of DNA Repair Systems and Involvement in Carcinogenesis
2.3. Interruption of Cell Growth Signaling and Its Promotion of Carcinogenesis
3. Revisiting Potential Biomarkers for Metal-Genotoxicity and Carcinogenicity: Interference of Protein-Protein Interactions with Zinc Finger Proteins
3.1. Mammalian DNA Repair Protein XPA
3.2. Poly (ADP-Ribose)Polymerase(PARP)
3.3. Tumor Suppressor Protein p53
4. Enhancement of Antioxidant Defense Systems Responsible for Reducing Metal-Induced Carcinogenicity
4.1. Enzymatic Antioxidants and Their Physiological Response to Metallotoxicity
4.2. Non-Enzymatic Antioxidants and Their Melioration towards Carcinogenesis
5. Conclusion
Acknowledgments
References
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol 2008, 82, 493–512. [Google Scholar]
- Hartwig, A. Zinc finger proteins as potential targets for toxic metal ions: Differential effects on structure and function. Antioxid. Redox Signal 2001, 3, 625–634. [Google Scholar]
- Nieboer, E.; Fletcher, G.G.; Thomassen, Y. Relevance of reactivity determinants to exposure assessment and biological monitoring of the elements. J. Environ. Monit 1999, 1, 1–14. [Google Scholar]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar]
- Matés, J.M.; Pérez-Gómez, C.; de Castro, I.N.; Asenjo, M.; Márquez, J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int. J. Biochem. Cell Biol 2002, 34, 439–458. [Google Scholar]
- Matés, J.M.; Segura, J.A.; Alonso, F.J.; Márquez, J. Intracellular redox status and oxidative stress: Implications for cell proliferation, apoptosis, and carcinogenesis. Arch. Toxicol 2008, 82, 273–299. [Google Scholar]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem 2004, 11, 1163–1182. [Google Scholar]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem 2004, 266, 37–56. [Google Scholar]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact 2006, 160, 1–40. [Google Scholar]
- Price, D.J.; Joshi, J.G. Ferritin. Binding of beryllium and other divalent metal ions. J. Biol. Chem 1983, 258, 10873–10880. [Google Scholar]
- Casalino, E.; Sblano, C.; Landriscina, C. Enzyme activity alteration by cadmium administration to rats: The possibility of iron involvement in lipid peroxidation. Arch. Biochem. Biophys 1997, 346, 171–179. [Google Scholar]
- Park, J.Y.; Seo, Y.R. The protective role of Nrf2 in cadmium-induced DNA damage. Mol. Cell. Toxicol 2011, 7, 61–66. [Google Scholar]
- Yang, J.M.; Arnush, M.; Chen, Q.Y.; Wu, X.D.; Pang, B.; Jiang, X.Z. Cadmium-induced damage to primary cultures of rat Leydig cells. Reprod. Toxicol 2003, 17, 553–560. [Google Scholar]
- Qu, W.; Diwan, B.A.; Reece, J.M.; Bortner, C.D.; Pi, J.; Liu, J.; Waalkes, M.P. Cadmium-induced malignant transformation in rat liver cells: Role of aberrant oncogene expression and minimal role of oxidative stress. Int. J. Cancer 2005, 114, 346–355. [Google Scholar]
- Witkiewicz-Kucharczyk, A.; Bal, W. Damage of zinc fingers in DNA repair proteins, a novel molecular mechanism in carcinogenesis. Toxicol. Lett 2006, 162, 29–42. [Google Scholar]
- Shukla, D.; Saxena, S.; Jayamurthy, P.; Sairam, M.; Singh, M.; Jain, S.K.; Bansal, A.; Ilavazaghan, G. Hypoxic preconditioning with cobalt attenuates hypobaric hypoxia-induced oxidative damage in rat lungs. High Alt. Med. Biol 2009, 10, 57–69. [Google Scholar]
- Genestra, M. Oxyl radicals, redox-sensitive signallingcascades and antioxidants. Cell. Signal 2007, 19, 1807–1819. [Google Scholar]
- Kim, D.-S.; Yu, S.-D.; Lee, E.-H. Effects of blood lead concentration on intelligence and personality in school children. Mol. Cell. Toxicol 2010, 6, 19–23. [Google Scholar]
- Clarkson, T.W.; Magos, L. The toxicology of mercury and its chemical compounds. Crit. Rev. Toxicol 2006, 36, 609–662. [Google Scholar]
- Kim, D.S.; Kim, J.H.; Yang, W.-H.; Moon, J.S.; Son, B.S. Biomonitoring of urinary mercury in Korean school children. Mol. Cell. Toxicol 2010, 6, 353–360. [Google Scholar]
- Peters, K.; Unger, R.E.; Gatti, A.M.; Sabbioni, E.; Tsaryk, R.; Kirkpatrick, C.J. Metallic nanoparticles exhibit paradoxical effects on oxidative stress and pro-inflammatory response in endothelial cells in vitro. Int. J. Immunopathol. Pharmacol 2007, 20, 685–695. [Google Scholar]
- Khalil, W.K.; Girgis, E.; Emam, A.N.; Mohamed, M.B.; Rao, K.V. Genotoxicity evaluation of nanomaterials: DNA damage, micronuclei, and 8-hydroxy-2-deoxyguanosine induced by magnetic doped CdSe quantum dots in male mice. Chem. Res. Toxicol 2011, 24, 640–650. [Google Scholar]
- Kim, J.-H.; Jang, A.-S.; Shin, E.K.; Kang, C.-M.; Seok, J.; Lee, E.H.; Kim, M.O.; Park, S.W.; Uh, S.T.; Park, C.-S. Particle-induced expression of SF20/IL25 is mediated by reactive oxygen species and NF-κB in alveolar macrophages. Mol. Cell. Toxicol 2010, 6, 305–312. [Google Scholar]
- Kim, Y.-J.; Yang, S.I.; Ryu, J.-C. Cytotoxicity and genotoxicity of nano-silver in mammalian cell lines. Mol. Cell. Toxicol 2010, 6, 119–125. [Google Scholar]
- Park, H.-O.; Yu, M.; Kang, S.K.; Yang, S.I.; Kim, Y.-J. Comparison of cellular effects of titanium dioxide nanoparticles with different photocatalytic potential in human keratinocyte, HaCaT cells. Mol. Cell. Toxicol 2011, 7, 67–75. [Google Scholar]
- Yeo, M.-K.; Kang, M.-S. The effect of nano-scale Zn-doped TiO2 and pure TiO2 particles on Hydra magnipapillata. Mol. Cell. Toxicol 2010, 6, 9–17. [Google Scholar]
- Jeon, Y.-M.; Park, S.-K.; Rhee, S.-K.; Lee, M.-Y. Proteomic profiling of the differentially expressed proteins by TiO2 nanoparticles in mouse kidney. Mol. Cell. Toxicol 2010, 6, 419–425. [Google Scholar]
- Yeo, M.-K.; Kim, H.-E. Gene expression in zebrafish embryos following exposure to TiO2 nanoparticles. Mol. Cell. Toxicol 2010, 6, 97–104. [Google Scholar]
- Yeo, M.K.; Park, S.W. Exposing zebrafish to silver nanoparticles during caudal fin regeneration disrupts caudal fin growth and p53 signaling. Mol. Cell. Toxicol 2008, 4, 311–317. [Google Scholar]
- Yeo, M.K.; Yoon, J.W. Comparison of the effects of nano-silver antibacterial coating and silver ions on zebrafish embryogenesis. Mol. Cell. Toxicol 2009, 5, 23–31. [Google Scholar]
- Hartwig, A.; Schwerdtle, T. Interactions by carcinogenic metal compounds with DNA repair processes: Toxicological implications. Toxicol. Lett 2002, 127, 47–54. [Google Scholar]
- Kasten, U.; Mullenders, L.H.; Hartwig, A. Cobalt(II) inhibits the incision and the polymerization step of nucleotide excision repair in human fibroblasts. Mutat. Res 1997, 383, 81–89. [Google Scholar]
- Hartwig, A.; Mullender, L.; Asmuβ, M.; Dally, H.; Hartmann, M. Disruptions of DNA repair processes by carcinogenic metal compounds. Fresenius J. Anal. Chem 1998, 361, 377–380. [Google Scholar]
- Calsoul, P.; Frit, P.; Bozzato, C.; Salles, B. Negative interference of metal(II) ions with nucleotide excision repair in human cell-free extracts. Carcinogenesis 1996, 17, 2779–2782. [Google Scholar]
- Schwerdtle, T.; Ebert, F.; Thuy, C.; Richter, C.; Mullenders, L.H.; Hartwig, A. Genotoxicity of soluble and particulate cadmium compounds: Impact on oxidative DNA damage and nucleotide excision repair. Chem. Res. Toxicol 2010, 23, 432–442. [Google Scholar]
- Hartwig, A.; Blessing, H.; Schwerdtle, T.; Walter, I. Modulation of DNA repair processes by arsenic and selenium compounds. Toxicology 2003a, 193, 161–169. [Google Scholar]
- Hartwig, A.; Pelzer, A.; Asmuss, M.; Bürkle, A. Very low concentrations of arsenite suppress poly(ADP-ribosyl)ation in mammalian cells. Int. J. Cancer 2003b, 104, 1–6. [Google Scholar]
- Schwerdtle, T.; Walter, I.; Hartwig, A. Arsenite and its biomethylated metabolites interfere with the formation and repair of stable BPDE-induced DNA adducts in human cells and impair XPAzf and Fpg. DNA Repair 2003, 2, 1449–1463. [Google Scholar]
- Hu, W.; Feng, Z.; Tang, M. Chromium(VI) enhances (±)-anti-7β,8α-dihydroxy-9α,10α-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene-induced cytotoxicity and mutagenicity in mammalian cells through its inhibitory effect on nucleotide excision repair. Biochemistry 2004, 43, 14282–14289. [Google Scholar]
- Lee, A.J.; Hodges, N.J.; Chipman, J.K. Interindividual variability in response to sodium dichromate-induced oxidative DNA damage: Role of the Ser326Cys polymorphism in the DNA repair protein of 8-oxo-7,8-dihydro-2′-deoxyguanosine DNA glycosylase 1. Cancer Epidermiol. Biomark. Prev 2005, 14, 497–505. [Google Scholar]
- Ford, B.N.; Ruttan, C.C.; Kyle, V.L.; Brackley, M.E.; Glickman, B.W. Identification of single nucleotide polymorphisms in human DNA repair genes. Carcinogenesis 2000, 21, 1977–1981. [Google Scholar]
- Nohmi, T.; Kim, S.R.; Yamada, M. Modulation of oxidative mutagenesis and carcinogenesis by polymorphic forms of human DNA repair enzymes. Mutat. Res 2005, 591, 60–73. [Google Scholar]
- Poli, G.; Leonarduzzi, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signalling. Curr. Med. Chem 2004, 11, 1163–1182. [Google Scholar]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol 2000, 279, L1005–L1028. [Google Scholar]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulations by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med 1996, 74, 589–607. [Google Scholar]
- Pinkus, R.; Weiner, L.M.; Daniel, V. Role of oxidants and antioxidants in the induction of AP-1, NF-kappa B, and glutathione S-transferase gene expression. J. Biol. Chem 1996, 271, 13422–13429. [Google Scholar]
- Amiri, K.I.; Richmond, A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev 2005, 24, 301–313. [Google Scholar]
- Pande, V.; Ramos, M.J. NF-kappa B in human disease: Current inhibitors and prospects for de novo structure based design of inhibitors. Curr. Med. Chem 2005, 12, 357–374. [Google Scholar]
- Knight, J.A. Free radicals, antioxidants, and the immune system. Ann. Clin. Lab. Sci 2000, 30, 145–158. [Google Scholar]
- Hughes, G.; Murphy, M.P.; Ledgerwood, E.C. Mitochondrial reactive oxygen species regulate the temporal activation of nuclear factor kappa B to modulate tumour necrosis factor-induced apoptosis: Evidence from mitochondria-targeted antioxidants. Biochem. J 2005, 389, 83–89. [Google Scholar]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancer. Science 1991, 253, 49–53. [Google Scholar]
- Maehle, L.; Metcalf, R.A.; Ryberg, D.; Bennett, W.P.; Harris, C.C.; Haugen, A. Altered p53 gene structure and expression in human epithelial cells after exposure to nickel. Cancer Res 1992, 52, 218–221. [Google Scholar]
- Olivier, M.; Hussain, S.P.; de Fromentel, C.C.; Hainaut, P.; Harris, C.C. TP53 mutation specta and load: A tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ 2004, 157, 247–270. [Google Scholar]
- Huang, C.; Ma, W.Y.; Li, J.; Dong, Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res 1999, 59, 3053–3058. [Google Scholar]
- Wang, S.W.; Shi, X.L. Mechanisms of Cr(VI)-induced p53 activation: The role of phosphorylation, mdm2 and ERK. Carcinogenesis 2001, 22, 757–762. [Google Scholar]
- Jauliac, S.; Lopez-Rodriguez, C.; Shaw, L.M.; Brown, L.F.; Rao, A.; Toker, A. The role of FAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol 2002, 4, 540–544. [Google Scholar]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Ann. Rev. Immunol 1997, 15, 707–747. [Google Scholar]
- Chow, C.W.; Rincon, M.; Cavanagh, J.; Dickens, M.; Davis, R.J. Nuclear accumulation of NFAT4 opposed by the JNK signal transduction pathway. Science 1997, 278, 1638–1641. [Google Scholar]
- Leonard, S.S.; Harris, G.K.; Shi, X.L. Metal-induced oxidative stress and signal transduction. Free Radic. Biol. Med 2004, 37, 1921–1942. [Google Scholar]
- Semenza, G.L. HIF-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol 2000, 88, 1474–1480. [Google Scholar]
- Gao, N.; Jiang, B.H.; Leonard, S.S.; Corum, L.; Zhang, Z.; Roberts, J.R.; Antonini, J.; Zheng, J.Z.; Flynn, D.C.; Castranova, V.; et al. p38 signaling-mediated hypoxia-inducible factor 1 alpha and vascular endothelial growth factor induction by Cr(VI) in DU145 human prostate carcinoma cells. J. Biol. Chem 2002, 277, 45041–45048. [Google Scholar]
- Salnikow, K.; Zhitkovich, A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol 2008, 21, 28–44. [Google Scholar]
- Maret, W.; Li, Y. Coordination dynamics of zinc in proteins. Chem. Rev 2009, 109, 4682–4707. [Google Scholar]
- Parkin, G. Synthetic analogues relevant to the structure and function of zinc enzymes. Chem. Rev 2004, 104, 699–767. [Google Scholar]
- Quintal, S.M.; de Paula, Q.A.; Farrell, N.P. Zinc finger proteins as templates for metal ion exchange and ligan reactivity. Chemical and biological consequences. Metallomics 2011, 2, 121–139. [Google Scholar]
- de Paula, Q.A.; Mangrum, J.B.; Farrell, N.P. Zinc finger proteins as templates for metal ion exchange: Substitution effects on the C-finger of HIV nucleocapsid NCp7 using M (chelate) species (M = Pt, Pd, Au). J. Inorg. Biochem 2009, 103, 1347–1354. [Google Scholar]
- Hartwig, A.; Asmuss, M.; Blessing, H.; Hoffmann, S.; Jahnke, G.; Khandelwal, S.; Pelzer, A.; Bürkle, A. Interference by toxic metal ions with zinc-dependent proteins involved in maintaining genomic stability. Food Chem. Toxicol 2002, 40, 1179–1184. [Google Scholar]
- Hartwig, A. Zinc finger proteins as potential targets for toxic metal ions: Differential effects on structure and function. Antioxid. Redox Signal 2001, 3, 625–634. [Google Scholar]
- Sunderman, F.W., Jr; Barber, A.M. Finger-loops, oncogenes, and metals. Claude Passmore Brown Memorial Lecture. Ann. Clin. Lab. Sci. 1988, 18, 267–288. [Google Scholar]
- Miyamoto, I.; Miura, N.; Niwa, H.; Miyazaki, J.; Tanaka, K. Mutational analysis of the structure and function of the xerodermapigmentosum group A complementing protein. J. Biol. Chem 1992, 267, 12182–12187. [Google Scholar]
- Riedl, T.; Hanaoka, F.; Egly, J.-M. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J 2003, 22, 5293–5303. [Google Scholar]
- Asahina, H.; Kuraoka, I.; Shirakawa, M.; Morita, E.H.; Miura, N.; Miyamoto, I.; Ohtsuka, E.; Okada, Y.; Tanaka, K. The XPA protein a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat. Res 1994, 315, 229–237. [Google Scholar]
- Jones, C.J.; Wood, R.D. Preferential binding of the xerodermapigmentosum group A complementing protein to damaged DNA. Biochemistry 1993, 32, 12096–12104. [Google Scholar]
- Robins, P.; Jones, C.J.; Biggerstaff, M.; Lindahl, T.; Wood, R.D. Complementation of DNA repair in xerodermapigmentosum group A cell extracts by a protein with affinity for damaged DNA. EMBO J 1991, 10, 3913–3921. [Google Scholar]
- Asmuss, M.; Mullenders, L.H.F.; Eker, A.; Hartwig, A. Differential effects of toxic metal compounds on the activities of Fpg and XPA, two zinc finger proteins involved in DNA repair. Carcinogenesis 2000, 21, 2097–2104. [Google Scholar]
- Bal, W.; Schwerdtle, T.; Hartwig, A. Mechanism of nickel assault on the zinc finger of DNA repair protein XPA. Chem. Res. Toxicol 2003, 16, 242–248. [Google Scholar]
- Kopera, E.; Schwerdtle, T.; Hartwig, A.; Bal, W. Co(II) and Cd(II) substitute for Zn(II) in the zinc finger derived from the DNA repair protein XPA, demonstrating a variety of potential mechanisms of toxicity. Chem. Res. Toxicol 2004, 17, 1452–1458. [Google Scholar]
- Gradwohl, F.; de Murcia, J.M.M.; Molinete, M.; Simonin, F.; Koken, M.; Hoeijmakers, J.H.; de Murcia, G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc. Natl. Acad. Sci. USA 1990, 87, 2990–2994. [Google Scholar]
- Kim, M.Y.; Zhang, T.; Kraus, W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD(+) into a nuclear signal. Genes Dev 2005, 19, 1951–1967. [Google Scholar]
- Homburg, S.; Visochek, L.; Moran, N.; Dantzer, F.; Priel, E.; Asculai, E.; Schwartz, D.; Rotter, V.; Dekel, N.; Cohen-Armon, M. A fast signal-induced activation of poly(ADP-ribose) polymerase: A novel downstream target of phospholipase C. J. Cell Biol 2000, 150, 293–307. [Google Scholar]
- Mendes, F.; Groessl, M.; Nazarov, A.A.; Tsybin, Y.O.; Sava, G.; Santos, I.; Dyson, P.J.; Casini, A. Metal-based inhibition of poly(ADP-ribose) polymerase-the guardian angel of DNA. J. Med. Chem 2011, 54, 2196–2206. [Google Scholar]
- de Murcia, G.; de Murcia, J.M. Poly(ADP-ribose) polymerase-A molecular nick-sensor. Trends Biochem. Sci 1994, 19, 172–176. [Google Scholar]
- Herceg, Z.; Wang, Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. Fundam. Mol. Mech. Mutagen 2001, 477, 97–110. [Google Scholar]
- de Murcia, G.; Shall, S. Poly(ADP-ribosylation) reactions: From DNA damage and stress signalling to cell death. Mutat. Res 2000, 460, 1–15. [Google Scholar]
- Yager, J.W.; Wiencke, J.K. Inhibition of poly(ADP-ribose) polymerase by arsenite. Mutat. Res 1997, 386, 345–351. [Google Scholar]
- Hainaut, P.; Mann, K. Zinc binding and redox control of p53 structure and function. Antioxid. Redox Signal 2001, 3, 611–623. [Google Scholar]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res 1999, 77, 81–137. [Google Scholar]
- Méplan, C.; Richard, M.J.; Hainaut, P. Redox signalling and transition metals in the control of p53 pathway. Biochem. Pharmacol 2000, 59, 25–33. [Google Scholar]
- Fischer, J.L.; Mihelc, E.M.; Pollok, K.E.; Smith, M.L. Chemotherapeutic selectivity conferred by selenium: A role for p53-dependent DNA repair. Mol. Cancer Ther 2007, 6, 355–361. [Google Scholar]
- Jung, H.J.; Seo, Y.R. Current issues of selenium in cancer chemoprevention. Biofactors 2010, 36, 153–158. [Google Scholar]
- Jung, H.J.; Lee, J.H.; Seo, Y.R. Enhancement of methylmethanesulfonate-induced based excision repair in the presence of selenomethionine on p53-dependent pathway. J. Med. Food 2009, 12, 340–344. [Google Scholar]
- Seo, Y.R.; Kelley, M.R.; Smith, M.L. Selenomethionine regulation of p53 by a ref 1-dependent redox mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 14548–14553. [Google Scholar]
- Seo, Y.R.; Sweeney, C.; Smith, M.L. Selenomethionine induction of DNA repair response in human fibroblasts. Oncogene 2002, 21, 3663–3669. [Google Scholar]
- Smith, M.L.; Lancia, J.K.; Mercer, T.I.; Ip, C. Selenium compounds regulate p53 by common and distinctive mechanisms. Anti-Cancer Res 2004, 24, 1401–1408. [Google Scholar]
- Palecek, E.; Brazdova, M.; Cemocka, H.; Vlk, D.; Brazda, V.; Vojtesek, B. Effect of transition metals on binding of p53 protein to supercoiled DNA and to consensus sequence in DNA fragments. Oncogene 1999, 18, 3617–3625. [Google Scholar]
- Meplan, C.; Mann, K.; Hainaut, P. Cadmium induces conformational modifications of wild-type p53 and suppress p53 response to DNA damage in cultured cells. J. Biol. Chem 1999, 274, 31663–31670. [Google Scholar]
- Cabelof, D.C.; Raffoul, J.J.; Yanamadala, S.; Ganir, C.; Guo, Z.M.; Heydari, A.R. Attenuation of DNA polymerase-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat. Res 2002, 500, 135–145. [Google Scholar]
- Koedrith, P.; Seo, Y.R. Development of quantitative DNA cleavage assay for XPG endonuclease activity using endogenous nuclear proteins in human cell lines. Oncol. Rep 2004, 279, 18425–18433. [Google Scholar]
- Mates, J.M.; Perez-Gomez, C.; de Castro, I.N. Antioxidant enzymes and human diseases. Clin. Biochem 1999, 32, 595–603. [Google Scholar]
- McCall, M.R.; Frei, B. Can antioxidant vitamins materially reduce oxidative damage in humans? Free Radic. Biol. Med 1999, 26, 1034–1053. [Google Scholar]
- Sies, H.; Stahl, W.; Sevanian, A. Nutritional, dietary and post-prandial oxidative stress. J. Nutr 2005, 135, 969–972. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M.C. Role of free radicals and catalytic metal-ions in human disease-an overview. Methods Enzymol 1990, 186, 1–85. [Google Scholar]
- Maiti, S.; Chatterjee, A.K. Effects on levels of glutathione and some related enzymes in tissues after an acute exposure in rats and their relationship to dietary protein deficiency. Arch. Toxicol 2001, 75, 531–537. [Google Scholar]
- Novelli, E.I.B.; Marques, S.F.G.; Almeida, J.A.; Diniz, Y.S.; Faine, L.A.; Ribas, B.O. Toxic mechanism of cadmium exposure on cardiac tissue. Toxic Subst. Mech 2000, 19, 207–217. [Google Scholar]
- Ognjanovic, B.I.; Pavlovic, S.Z.; Maletic, S.D.; Zikic, R.V.; Stajn, A.S.; Radojicic, R.M.; Saicic, Z.S.; Petrovic, V.M. Protective influence of vitamin E on antioxidant defense system in the blood of rats treated with cadmium. Physiol. Res 2003, 52, 563–570. [Google Scholar]
- Ahamed, M.; Verma, S.; Kumar, A.; Siddiqui, M.K.J. Environmental exposure to lead and its correlation with biochemical indices in children. Sci. Total Environ 2005, 346, 48–55. [Google Scholar]
- Hoffman, D.J.; Heinz, G.H.; Sileo, L.; Audet, D.J.; Campbell, J.K.; Obrecht, H.H. Developmental toxicity of lead-contaminated sediment in Canada geese (Branta canadensis). J. Toxicol. Environ. Health A 2000, 59, 235–252. [Google Scholar]
- Crespo-López, M.E.; Macêdo, G.L.; Pereira, S.I.D.; Arrifano, G.P.F.; Picanço-Diniz, D.L.W.; do Nascimento, J.L.M.; Herculano, A.M. Mercury and human genotoxicity: Critical considerations and possible molecular mechanisms. Pharmacol. Res 2009, 60, 212–220. [Google Scholar]
- Lee, C.-H.; Lin, R.-H.; Liu, S.H.; Lin-Shiau, S.-Y. Distinct genotoxicity of phenylmercury acetate in human lymphocytes as compared with other mercury compounds. Mutat. Res 1997, 392, 269–276. [Google Scholar]
- Pinheiro, M.C.N.; Macchi, B.M.; Vieira, J.L.F.; Oikawa, T.; Amoras, W.W.; Guimarães, G.A.; Costa, C.A.; Crespo-López, M.E.; Herculano, A.M.; Silveira, L.C.L.; et al. Mercury exposure and antioxidant defenses in women: A comparative study in the Amazon. Environ. Res 2008, 107, 53–59. [Google Scholar]
- Gupta, R.S.; Gupta, E.S.; Dhakal, B.K.; Thakur, A.R.; Ahnn, J. Vitamin C and vitamin E protect the rat testes from cadmium-induced reactive oxygen species. Mol. Cells 2004, 17, 132–139. [Google Scholar]
- Karbownik, M.; Gitto, E.; Lewinski, A.; Reiter, R.J. Induction of lipid peroxidation in hamster organs by the carcinogen cadmium: Melioration by melatonin. Cell Biol. Toxicol 2001, 17, 33–40. [Google Scholar]
- Cha, S.H.; Suh, C.K. Heme oxygenase-1 mediated protective effect of methyl gallate on cadmium-induced cytotoxicity in cultured mouse mesangial cells. Mol. Cell. Toxicol 2010, 6, 127–133. [Google Scholar]
- Park, M.S.; Shin, H.S.; Lee, J.H.; Kil, G.-S.; Choi, C.Y. Influence of quercetin on the physiological response to cadmium stress in olive flounder, Paralichthysolivaceus: Effects on hematological and biochemical parameters. Mol. Cell. Toxicol 2010, 6, 151–159. [Google Scholar]
- Fenoglio, I.; Corazzari, I.; Francia, C.; Bodoardo, S.; Fubini, B. The oxidation of glutathione by cobalt/tungsten carbide contributes to hard metal-induced oxidative stress. Free Radic. Res 2008, 42, 437–745. [Google Scholar]
- Salnikow, K.; Donald, S.P.; Bruick, R.K.; Zhitkovich, A.; Phang, J.M.; Kasprzak, K.S. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J. Biol. Chem 2004, 279, 40337–40344. [Google Scholar]
- Stefaniak, A.B.; Harvey, C.J.; Bukowski, V.C.; Leonard, S.S. Comparison of free radical generation by pre- and post-sintered cemented carbide particles. J. Occup. Environ. Hyg 2010, 7, 23–34. [Google Scholar]
- O’Brien, T.; Mandel, H.G.; Pritchard, D.E.; Patierno, S.R. Critical role of chromium (Cr)-DNA interactions in the formation of Cr-induced polymerase arresting lesions. Biochemistry 2002, 41, 12529–12537. [Google Scholar]
- Quievryn, G.; Messer, J.; Zhitkovich, A. Carcinogenic chromium(VI) induces cross-linking of vitamin C to DNA in vitro and in human lung 549 cells. Biochemistry 2002, 41, 3156–3167. [Google Scholar]
- Quievryn, G.; Peterson, E.; Messer, J.; Zhitkovich, A. Genotoxicity and mutagenicity of chromium(VI)/ascorbate-generated DNA adducts in human and bacterial cells. Biochemistry 2003, 42, 1062–1070. [Google Scholar]
- de Flora, S.; Wetterhahn, K.E. Mechanisms of chromium metabolism and genotoxicity. Life Chem. Rep 1989, 7, 169–244. [Google Scholar]
- Hunaiti, A.A.; Soud, M. Effect of lead concentration on the level of glutathione, glutathione S-transferase, reductase and peroxidase in human blood. Sci. Total Environ 2000, 248, 45–50. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Batra, N.; Nehru, B.; Bansal, M.P. The effect of zinc supplementation on the effects of lead in the rat testis. Reprod. Toxicol 1998, 12, 535–540. [Google Scholar]
- Othman, A.I.; El Missiry, M.A. Role of selenium against lead toxicity in male rats. J. Biochem. Mol. Toxicol 1998, 12, 345–349. [Google Scholar]
- Pande, M.; Flora, S.J.S. Lead induced oxidative damage and its response to combined administration of alpha-lipoic acid and succimers in rats. Toxicology 2002, 177, 187–196. [Google Scholar]
- Schurz, F.; Sabater-Vilar, M.; Fink-Gremmels, J. Mutagenicity of mercury chloride and mechanisms of cellular defence: The role of metal-binding proteins. Mutagenesis 2000, 15, 525–530. [Google Scholar]
- Herculano, A.M.; Crespo-Lopez, M.E.; Lima, S.M.A.; Pincanço-Diniz, D.L.W.; do Nascimento, J.L.M. Methylmercury intoxication activates nitric oxide synthase in chick retinal cell culture. Braz. J. Med. Biol. Res 2006, 39, 415–418. [Google Scholar]
- Rao, M.V.; Chinoy, N.J.; Suthar, M.B.; Rajvanshi, M.I. Role of ascorbic acid on mercuric chloride-induced genotoxicity in human blood cultures. Toxicol. In Vitro 2001, 15, 649–654. [Google Scholar]
- Passos, C.J.S.; Mergler, D.; Fillion, M.; Lemire, M.; Mertens, F.; Guimarães, J.R.D.; Philibert, A. Epidemiologic confirmation that fruit consumption influences mercury exposure in riparian communities in the Brazilian Amazon. Environ. Res 2007, 105, 183–193. [Google Scholar]
- Myung, S.-K.; Kim, Y.; Ju, W.; Choi, H.J.; Bae, W.K. Effects of antioxidant supplements on cancer prevention: Meta-analysis of randomized controlled trials. Ann. Oncol 2010, 21, 166–179. [Google Scholar]
- Mahaffey, K.R. Mercury exposure: Medical and public health issues. Trans. Am. Clin. Climatol. Assoc 2005, 116, 127–154. [Google Scholar]
- Rojas, M.; Seijas, D.; Agreda, O.; Rodríquez, M. Biological monitoring of mercury exposure in individuals referred to a toxicological center in Venezuela. Sci. Total Environ 2006, 354, 278–285. [Google Scholar]
- Ronchetti, R.; Zuurbier, M.; Jesenak, M.; Koppe, J.G.; Ahmed, U.F.; Ceccatelli, S.; Villa, M.P. Children’s health and mercury exposure. Acta Paediatr. Suppl 2006, 95, 36–44. [Google Scholar]
- Tchounwou, P.B.; Ayensu, W.K.; Ninashvili, N.; Sutton, D. Environmental exposure to mercury and its toxicopathologic implications for public health. Environ. Toxicol 2003, 18, 149–175. [Google Scholar]
- Ho, E. Zinc deficiency, DNA damage and cancer risk. J. Nutr. Biochem 2004, 15, 572–578. [Google Scholar]

© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koedrith, P.; Seo, Y.R. Advances in Carcinogenic Metal Toxicity and Potential Molecular Markers. Int. J. Mol. Sci. 2011, 12, 9576-9595. https://doi.org/10.3390/ijms12129576
Koedrith P, Seo YR. Advances in Carcinogenic Metal Toxicity and Potential Molecular Markers. International Journal of Molecular Sciences. 2011; 12(12):9576-9595. https://doi.org/10.3390/ijms12129576
Chicago/Turabian StyleKoedrith, Preeyaporn, and Young Rok Seo. 2011. "Advances in Carcinogenic Metal Toxicity and Potential Molecular Markers" International Journal of Molecular Sciences 12, no. 12: 9576-9595. https://doi.org/10.3390/ijms12129576