1. Introduction
The chemistry of metal carbonyl complexes of acyclic hydrocarbons dates back to the 1930 discovery by Reihlen
et al. [
1] of the mononuclear butadiene iron tricarbonyl complex, C
4H
6Fe(CO)
3 by the reaction of butadiene with iron pentacarbonyl at elevated temperatures. The proposed tetrahapto bonding of the butadiene ligand to the Fe(CO)
3 unit in this complex was confirmed in 1963 by Mills and Robinson [
2] using X-ray crystallography at −40 °C (
Figure 1A). In addition, in 1962 Murdoch and Weiss [
3] used the reaction of butadiene with Fe
2(CO)
9 at room temperature to synthesize the tetracarbonyl (η
2-C
4H
6)Fe(CO)
4 in which only one of the two C═C double bonds of the butadiene ligand is bonded to the iron atom. An additional product from the latter reaction was the binuclear complex C
4H
6[Fe(CO)
4]
2 in which each C═C double bond of the butadiene ligand is bonded to a separate Fe(CO)
4 unit with the iron atoms much too far apart for any kind of direct iron-iron bond. However, no binuclear butadiene iron carbonyl derivatives with short iron-iron distances suggesting iron-iron bonds have been synthesized. In order to assess the possibilities for binuclear iron carbonyl derivatives with iron-iron bonds we have performed a density functional theory (DFT) study on possible structures for (η
4-C
4H
6)
2Fe
2(CO)
n (
n = 5, 4, 3), predicted to have structures with formal Fe–Fe single bonds, Fe═Fe double bonds, and Fe≡Fe triple bonds, respectively [
4]. In general, the lowest energy structures for these (η
4-C
4H
6)
2Fe
2(CO)
n derivatives were found to be coaxial structures in which each metal atom is bonded to a single butadiene ligand (
Figure 1B).
This paper reports a DFT study of the still more highly unsaturated (C
4H
6)
2Fe
2(CO)
n (
n = 2, 1) derivatives. Such systems are interesting since strict adherence to the 18-electron rule suggests that these highly unsaturated derivatives might provide examples of very short formal iron-iron quadruple and quintuple bonds. However, for such systems containing two or fewer carbonyl groups, alternative perpendicular structures are possible in which the butadiene ligands bridge the pair of iron atoms (
Figure 1C). Structures of both types were found in this work. Quintet spin state structures were found to be the lowest energy structures for both (C
4H
6)
2Fe
2(CO)
2 and (C
4H
6)
2Fe
2(CO). In addition, interesting structures were found with agostic hydrogen atoms bridging Fe–C bonds.
2. Theoretical Methods
Density functional theory (DFT) including electron correlation effects has been shown to be a powerful and effective computational tool in organotransition metal chemistry [
5–
19]. Three DFT methods were used for this work. The first functional was BP86, which combines Becke’s 1988 exchange functional (B) with Perdew’s 1986 gradient corrected correlation functional method (P86), and usually provides better vibrational frequencies [
20,
21]. The second DFT method was B3LYP, which is the hybrid HF/DFT functional using the combination of the three-parameter Becke functional (B3) [
22] with the Lee-Yang-Parr (LYP) generalized gradient correlation functional [
23]. The third was a hybrid meta-GGA DFT method, M06-L, developed by Truhlar’s group [
24]. Recently Truhlar’s group made much progress to the development of improved exchange-correlation functionals that are essential for expanding the applicability of Kohn–Sham DFT, such as the M06 suite. Thus M06-L was constructed using three strategies: constraint satisfaction, modeling the exchange-correlation hole, and empirical fits. They concluded that M06-L is one of the best functionals for the study of organometallic and inorganic thermochemistry, and is the best functional for transition metal energetics. In comparing the first two DFT methods Reiher and collaborators found that B3LYP always overestimates the energy of high-spin states and BP86 overestimates the energies of low-spin states for a series of the Fe(II)-S complexes [
25]. In the present study, we found that the M06-L method predicts an intermediate energy difference, anticipated to be closer to the experimental. We therefore adopt the energy order predicted by the M06-L method, but list the BP86 and B3LYP results in the Supporting Information.
Basis sets have been chosen to provide continuity with a body of existing research on organometallic compounds. Fortunately, DFT methods are far less basis set sensitive than higher-level methods such as coupled cluster theory. In this work, the double-ζ plus polarization (DZP) basis sets used for C and O add one set of pure spherical harmonic d functions with orbital exponents α
d(C) = 0.75 and α
d(O) = 0.85 to the Huzinaga Dunning standard contracted DZ sets and are denoted as (9s5p1d/4s2p1d) [
26,
27]. For H, a set of p polarization functions α
p(H) = 0.75 is added to the Huzinaga Dunning DZ sets. For Fe, in our loosely contracted DZP basis set, the Wachters’ primitive set [
28] is used after being augmented by two sets of p functions and one set of d functions and then contracted using the method of Hood, Pitzer, and Schaefer [
29]. This basis set is denoted as (14s11p6d/10s8p3d).
The geometries of the structures were fully optimized using the Gaussian09 program [
30] with the three selected DFT methods and with the indicated DZP basis set. The vibrational frequencies were determined by evaluating analytically the second derivatives of the energy with respect to the nuclear coordinates at the same levels. The corresponding infrared intensities were also evaluated analytically. The fine grid (75, 302) was the default for evaluating integrals numerically, and the tight designation was the default for the energy convergence, as well as the tight option for the geometry optimizations. In some cases, the finer grid (120, 974) was used for investigating small imaginary vibrational frequencies. Natural bond orbital (NBO) analyses [
31] used the DZP BP86 method with the NBO 3.1 version attached in the Gaussian03 program.
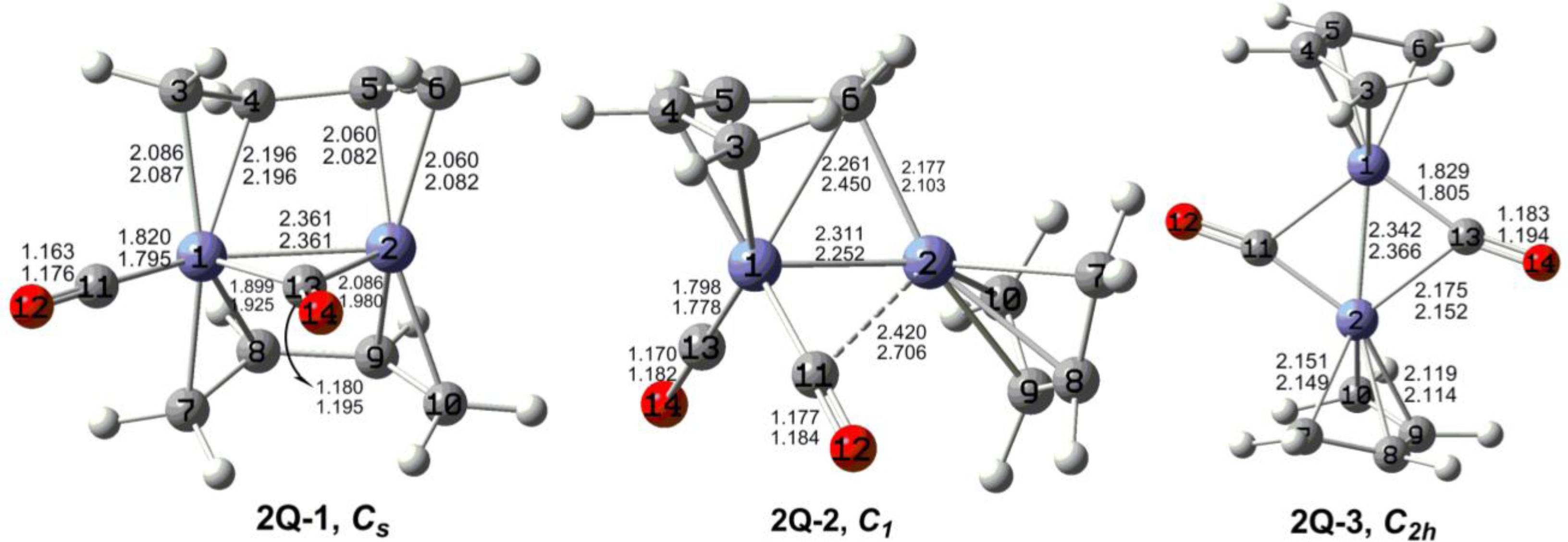
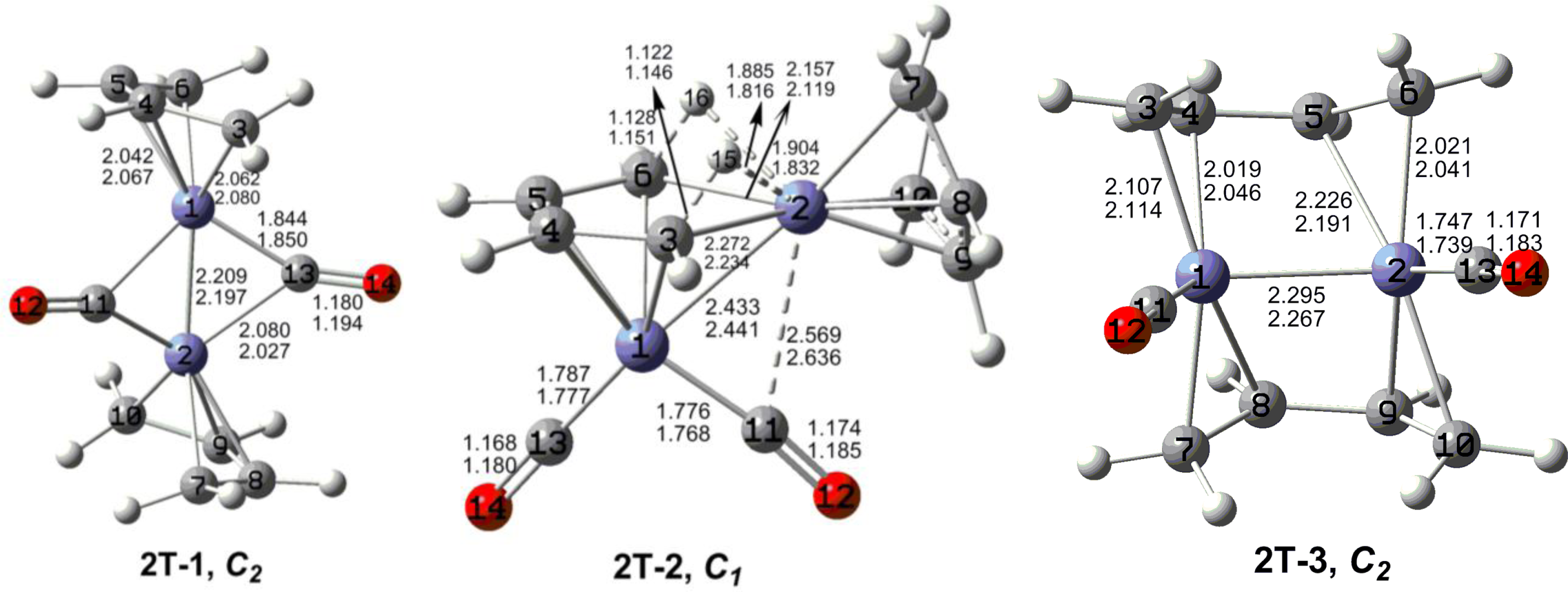
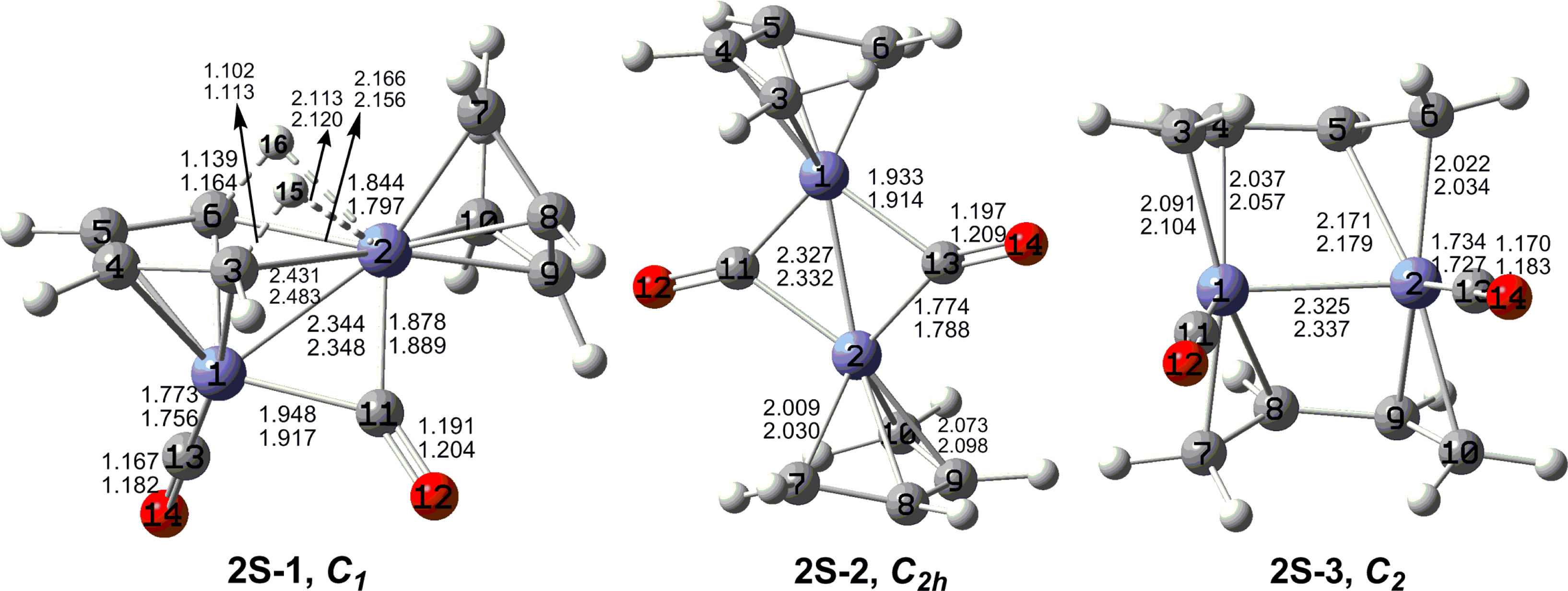
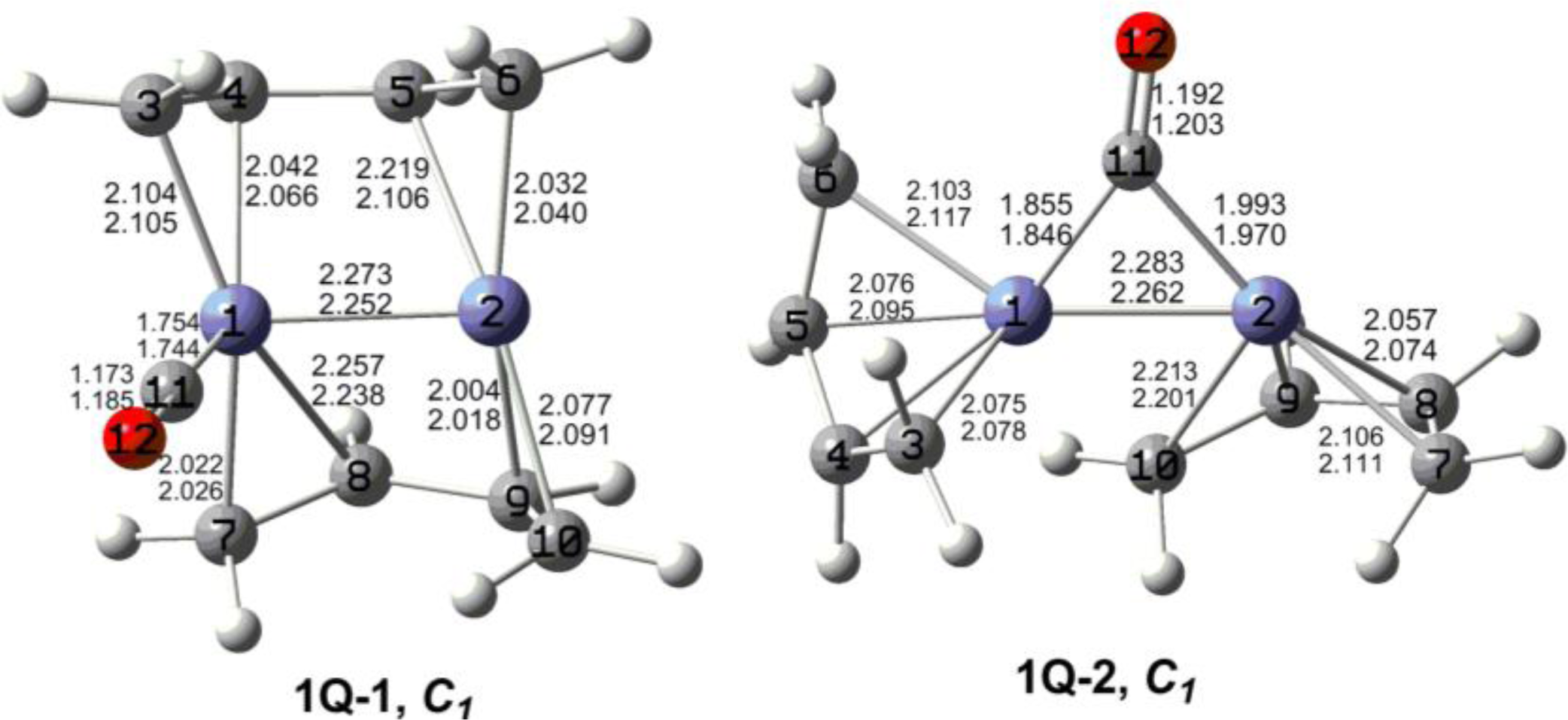
The optimized (C
4H
6)
2Fe
2(CO)
n (
n = 2, 1) structures are depicted in
Figures 2 to
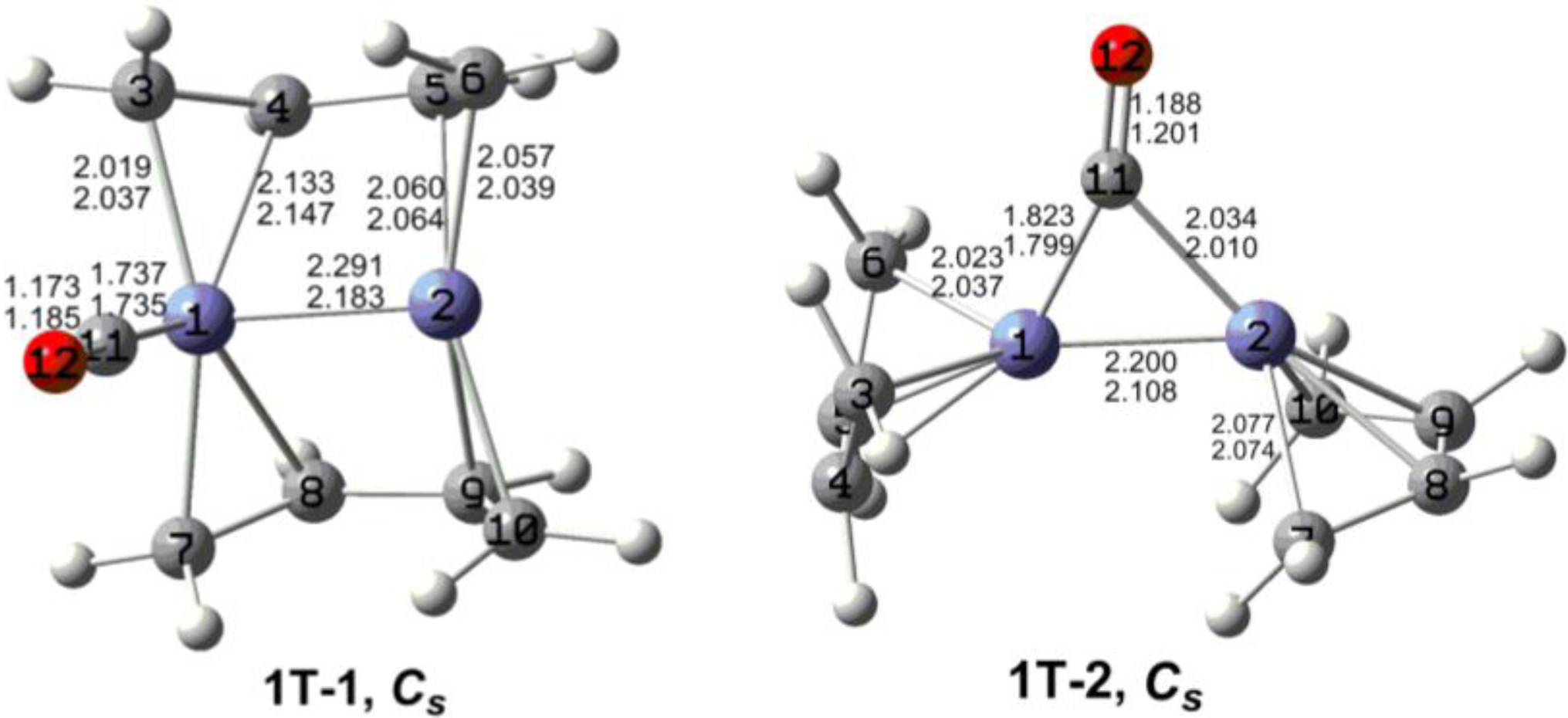
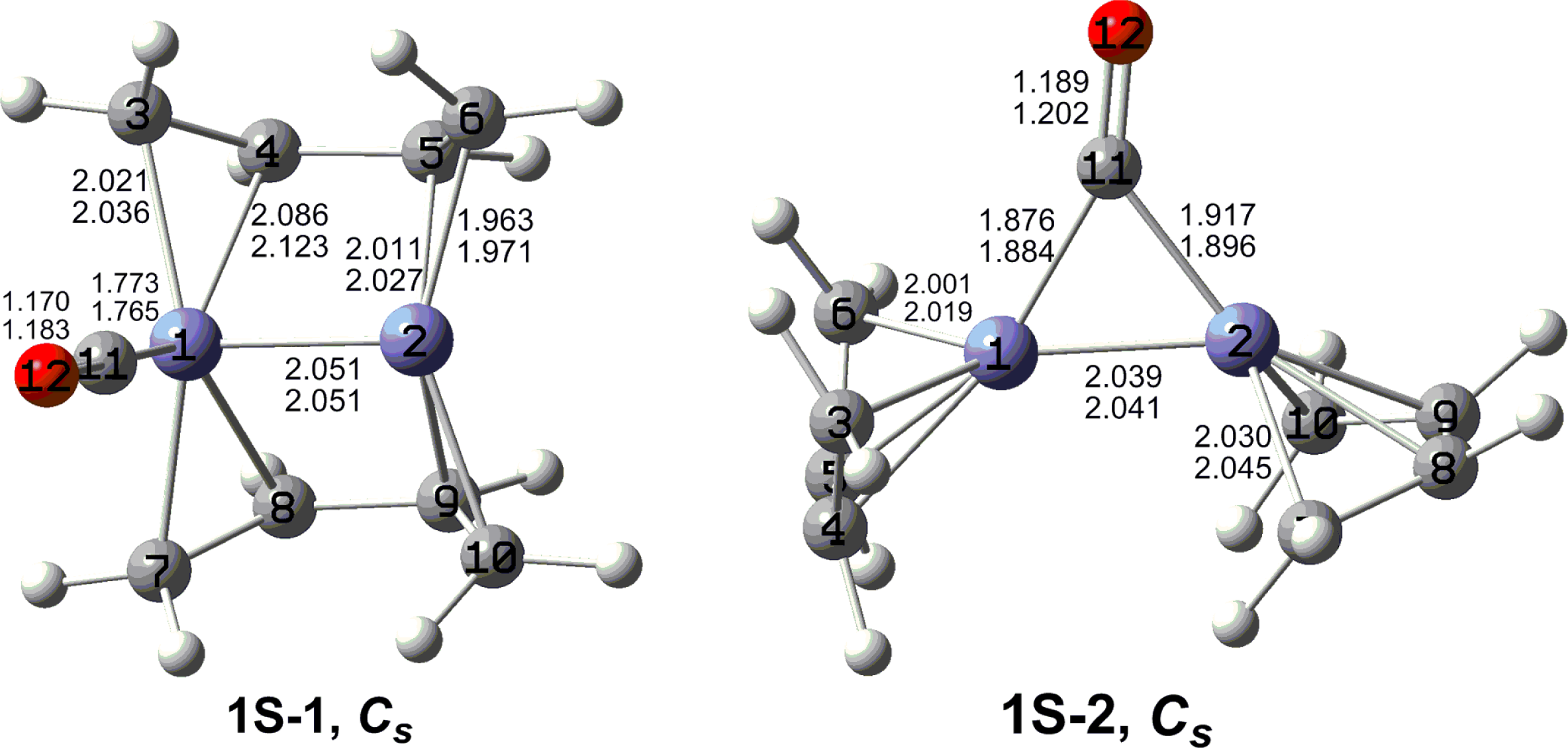
7. In these figures, the upper and lower distances were obtained by the M06-L and BP86 method, respectively. The structures are designated as
nX-m, where
n stands for the number of CO groups,
X designates the spin state using
S for singlets,
T for triplets and
Q for quintets, and
m orders the structures according to their relative energies. Note that, although the singlets, the triplets and the quintets are discussed in separate sections, the relative energies are considered together on the basis of the number of carbonyls. The M06-L method appears to predict the better singlet-triplet splittings.
The relative energies corrected for zero-point energies are listed in the Supporting Information, where computed enthalpies and free energies are also given. These relative free energies agree within 2 kcal/mol with the relative electronic energies.
4. Conclusions
Unsaturation in binuclear metal carbonyl derivatives can lead to metal-metal multiple bonding, four-electron donor bridging carbonyl groups, and/or metal electronic configurations less than the favorable 18-electron configurations. None of the highly unsaturated (C4H6)2Fe2(CO)n (n = 2, 1) structures found in this work has a four-electron donor bridging carbonyl group. Instead the lowest energy (C4H6)2Fe2(CO)n (n = 2, 1) structures are perpendicular structures having iron atoms with 15- and 17-electron configurations. This leads to quintet spin states in addition to iron-iron multiple bonds of formal order two for (C4H6)2Fe2(CO)2 and three for (C4H6)2Fe2(CO). In addition, agostic hydrogen atoms forming C-H-Fe bridges are seen to be a feature of (C4H6)2Fe2(CO)n (n = 2, 1) structures, albeit not the lowest energy such structures. In such structures a butadiene ligand is bonded to one of the iron atoms as a tetrahapto ligand and to the other iron atom through two C–H–Fe units from the end CH2 groups with agostic hydrogen atoms bridging iron-carbon bonds. Singlet (C4H6)2Fe2(CO) structures with formal Fe–Fe quadruple bonds of lengths ∼2.05 Å were also found but at very high energies (∼47 kcal/mol) relative to the global minimum.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}