2.1. Effects of Mutations on PARN Activity and Structure
All of the four recombinant mutated proteins were eluted at the same volume as the WT in the size-exclusion chromatography (SEC) profile (data not shown), indicating that the mutations of the four conserved acidic residues, D28, E30, D292 and D382, did not affect the oligomeric state of PARN. To investigate the effect of mutation on PARN function, the deadenylase activity was assayed by the methylene blue method [
19]. Consistent with the previous observations [
18], all four of the mutants were found to be completely inactive, suggesting that these four conserved acidic residues were essential for the two-metal-ion catalysis of PARN.
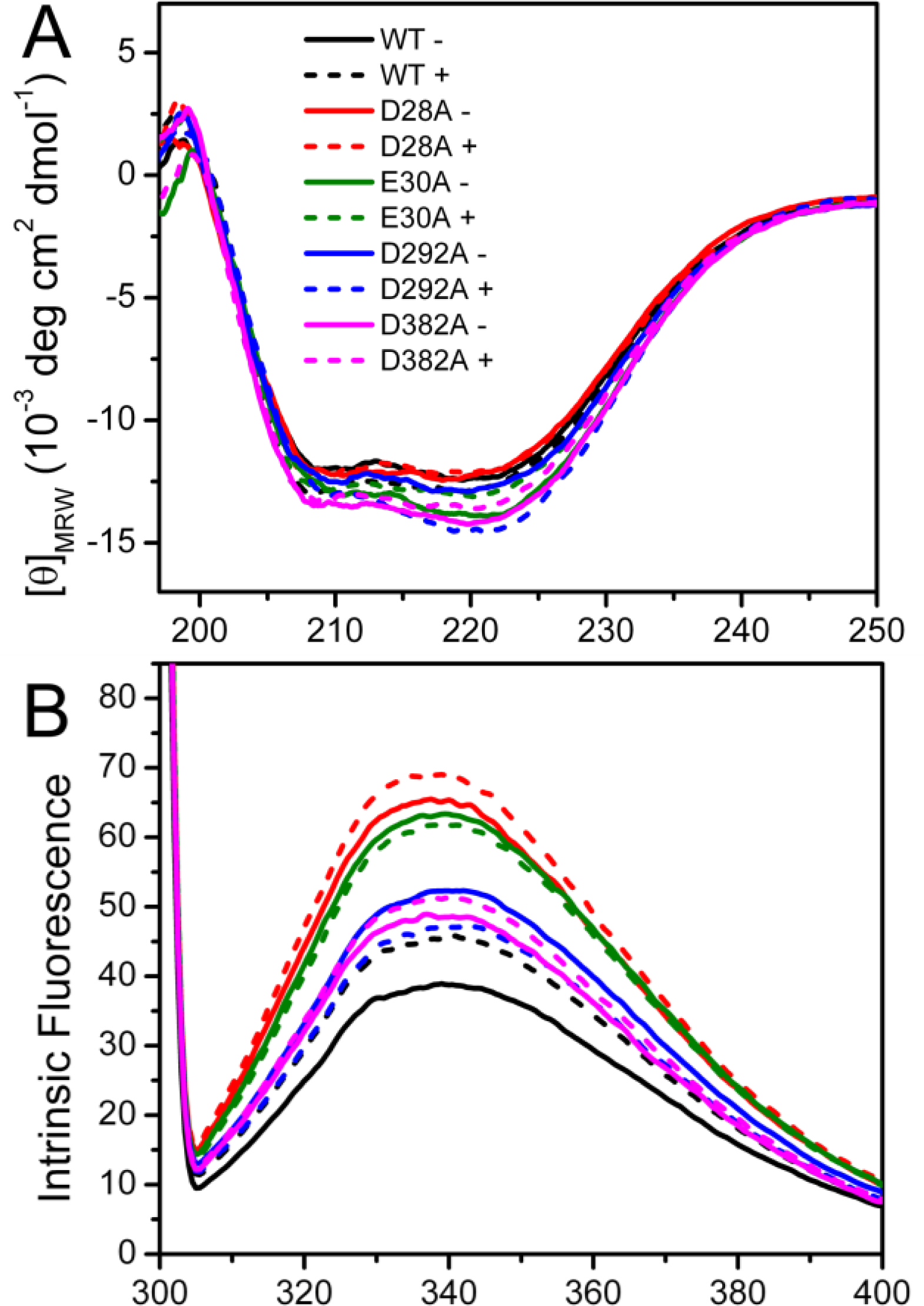
Spectroscopic experiments were carried out to assess the effect of mutation on PARN structure. The circular dichroism (CD) data shown in
Figure 2A indicated that the effects of the mutations on the secondary structure contents of PARN were minor. The four mutations affect PARN secondary structures dissimilarly: No significant changes were observed for the D28A and D292A mutations, while there was an ∼10% increase in the absolute value of ellipticity at 222 nm ([
θ222]) for the E30A and D382A mutations. With the addition of 3 mM Mg
2+, a minor increase of [
θ222] was observed for the WT protein. As for the mutants, the [
θ222] value changed little for D28A; there was a slight decrease for D382A and a significant decrease to the value close to that of the WT protein for E30A. However an ∼15% increase was found for D292A. These results indicated that the mutations either slightly increased or did not affect the percentages of the regular secondary structure contents of PARN. The coordination of Mg
2+ could rescue the disturbance in secondary structures induced by most mutations except D292A.
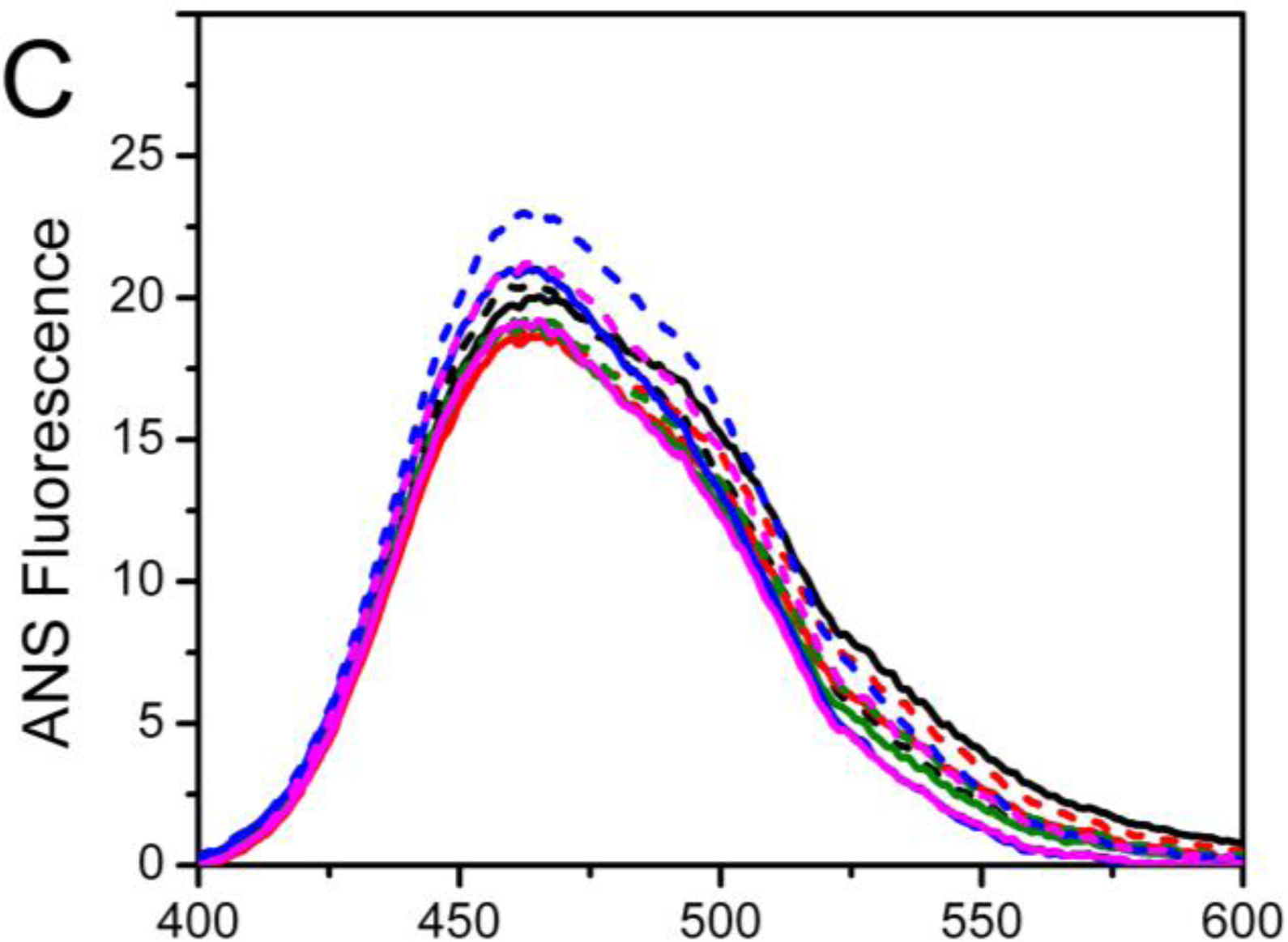
Intrinsic (
Figure 2B) and 1-anilinonaphtalene-8-sulfonate (ANS) fluorescence (
Figure 2C) were used to probe the effects of mutations on the PARN tertiary structure. When excited at 295 nm, the intrinsic fluorescence mainly reflects the microenvironmental status of Trp fluorophores. Consistent with previous observations [
20], the wavelength of the maximum Trp fluorescence was around 340 nm for both of the samples with and without Mg
2+. The mutations did not alter the maximum Trp fluorescence wavelength or shape of the spectra, suggesting that the mutations had no significant effect on the solvent accessibility of PARN Trp residues. Interestingly, the intrinsic fluorescence intensity was significantly increased due to the mutations. Because many factors may affect the fluorescence yield, it is difficult to determine accurately the reason for the intensity changes of intrinsic fluorescence. However, it is clear that the cofactor binding or mutation induced changes in the microenvironments around Trp residues.
Among the four mutants, the D28A and E30A mutations resulted in an ∼2-fold increase in Trp fluorescence, while the D292A and D382A mutations led to an ∼30% increase. With the addition of Mg2+, an increase in the Trp fluorescence intensity was observed for the WT protein (17%), D28A (6%) and D382A (5%); there was a slight quenching for E30A (3%) and D292A (10%). Meanwhile, the ANS fluorescence indicated that most of the mutants possessed similar ANS-binding ability to the WT PARN under both Mg2+-free and Mg2+-bound conditions, except that the Mg2+-bound D292A had a relative larger hydrophobic exposure.
Taken together, the minor difference in the CD signal, maximum Trp fluorescence wavelength and ANS fluorescence suggested that the mutations slightly modified PARN secondary and tertiary structures. The four mutations had dissimilar effects on PARN structure. Among them, D28A and E30A had similar secondary structures and hydrophobic exposure to the WT but great increase in Trp fluorescence intensity, while the D292A and D382A mutations resulted in the formation of more regular secondary structures, moderate increase in Trp fluorescence intensity, and different behavior upon Mg2+ coordination compared with the WT protein.
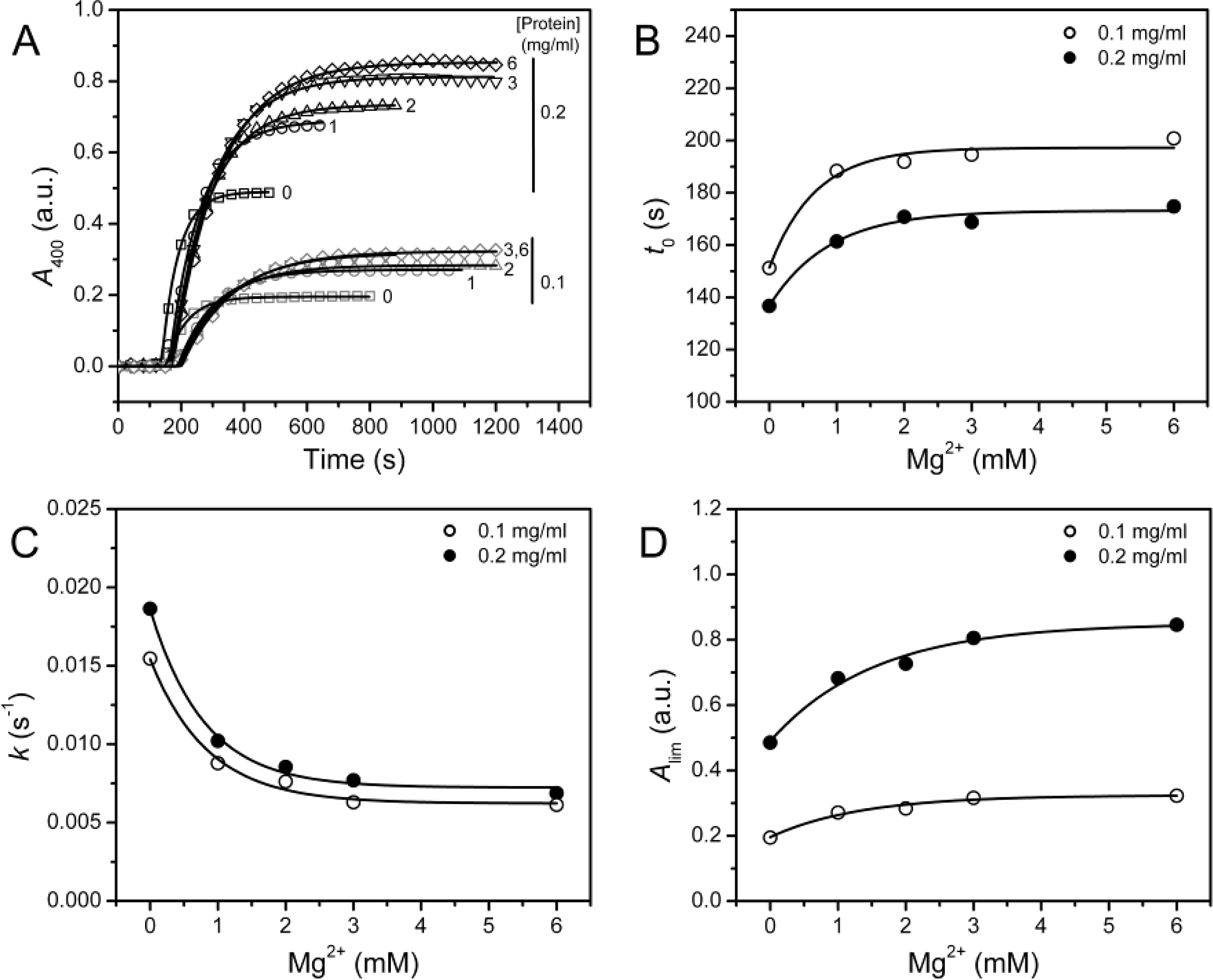
2.2. Effect of Mg2+ on PARN Thermal Aggregation
In a previous study, we found that 3 mM Mg
2+ can protect PARN against thermal inactivation below 60 °C, but it accelerates protein thermal aggregation at 61 °C [
12]. A similar result was observed when PARN was heated at 55 °C (
Figure 3A), where PARN thermal aggregation revealed Mg
2+- and protein-concentration dependent manner. The aggregation kinetics was analyzed by considering the protein aggregation process as an
n-th order reaction, and the following equation could be used for the fitting of the data [
21]
where
t is the time of incubation at a given temperature,
Alim is the
A400 value at the infinite time and
kn is the rate constant of the
n-th order reaction. Under all conditions, the aggregation was found to be dominated by a first-order kinetics with
n = 1.17 ± 0.08. To facilitate the comparison of the parameters, all the data were fitted by assuming
n = 1. The results shown in
Figure 3 indicated that all the three parameters (
t0,
k and
Alim) were significantly affected by the addition of Mg
2+ (
F test,
P < 0.0001). Unexpectedly, the three parameters revealed inconsistent results with the increase of [Mg
2+]: The increase in
t0 and the decrease in
k implied that Mg
2+ could inhibit the rate of PARN aggregation, whereas the increase in
Alim suggested that Mg
2+ increased the amounts or size of aggregates.
Turbidity, which was reflected by the absorbance at 400 nm in this research, is an indicator of the size of the aggregates [
22]. Since most proteins were found to exist in the aggregates after heat treatment, which were evidenced by the lack of soluble fractions when assayed by either spectroscopic methods or SDS-PAGE (data not shown), the increase of
Alim suggested that the protein might aggregate into larger forms in the presence of Mg
2+. Because the inactivation of PARN was much faster than its aggregation, the observations herein might explain the different effects of Mg
2+ on PARN inactivation and aggregation observed previously [
12]. The results shown in
Figure 3 also suggested that great caution should be taken when using
Alim alone to evaluate protein aggregation. To confirm the results in
Figure 3, the aggregation of PARN was also performed at 45 °C, and similar results were obtained except that aggregation process was significantly slowed down by the decrease of temperature (data not shown, see also
Figure 4). Thus both the variations in the protein concentration and incubating temperature did not alter the Mg
2+-dependence of PARN thermal aggregation.
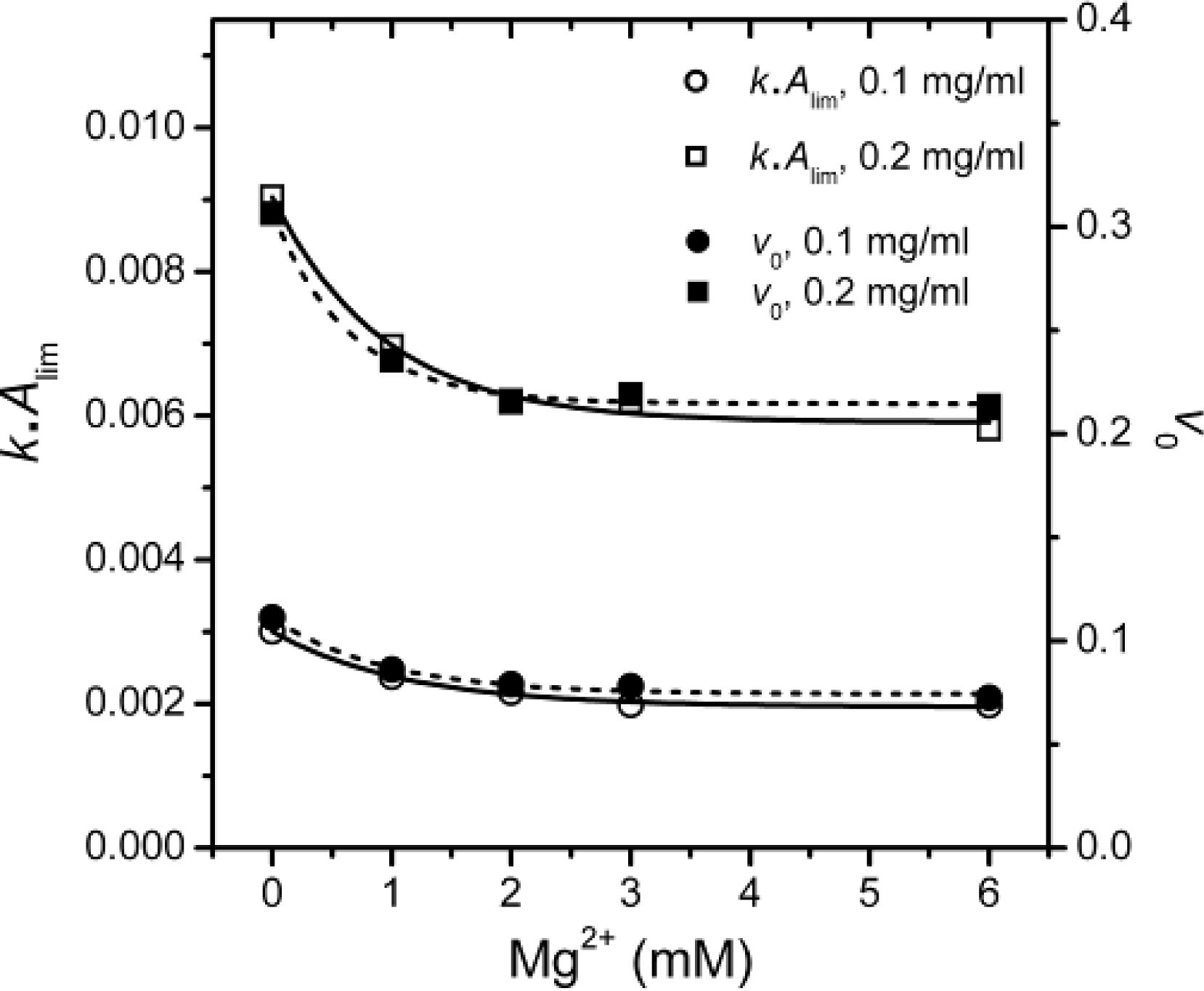
The inconsistency between
k and
Alim shown in
Figure 3 was also observed in the study of chaperone functions [
21]. It has been suggested that the value
kAlim, which reflects the initial velocity of aggregation, is a better indicator to measure the effects of cosolvents on first-order protein aggregation [
21]. The Mg
2+-dependence of the
kAlim values as well as the initial velocities of aggregation (
v0) were calculated and presented in
Figure 5.
It is clear that the
kAlim and
v0 values coincided with each other, and the results also indicated that Mg
2+ could decrease PARN thermal aggregation in a concentration dependent manner. The Mg
2+ dependence for the
kAlim data could be regarded as ligand-binding induced protein aggregation. Thus, the data in
Figure 5 could be fitted by the following equation
where Δ
Y is the relative change of the
kAlim value, and
appKD is the apparent dissociation constants of the aggregate·ligand complex. It is worth noting that the
appKD value only reflects the Mg
2+-dependence for PARN aggregation, but is not directly related to the binding of Mg
2+ to the aggregates. The
appKD values were 0.9 ± 0.2 and 0.7 ± 0.1 mM for 0.1 and 0.2 mg/mL PARN aggregation, respectively. Similar values could also be obtained from the data shown in
Figures 3B–3D. The small
appKD values of the WT PARN indicated the aggregation of the WT protein had a strong dependence on the existence of Mg
2+. This value is close to the optimal Mg
2+ concentration required for the catalysis of the 54 kDa PARN (∼1 mM) [
23], suggesting that the effects of Mg
2+ on PARN aggregation was related to Mg
2+-coordination in the active site.
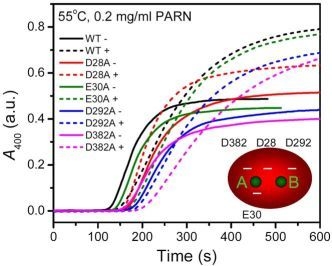
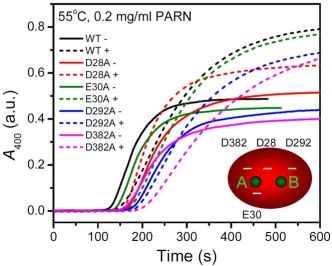
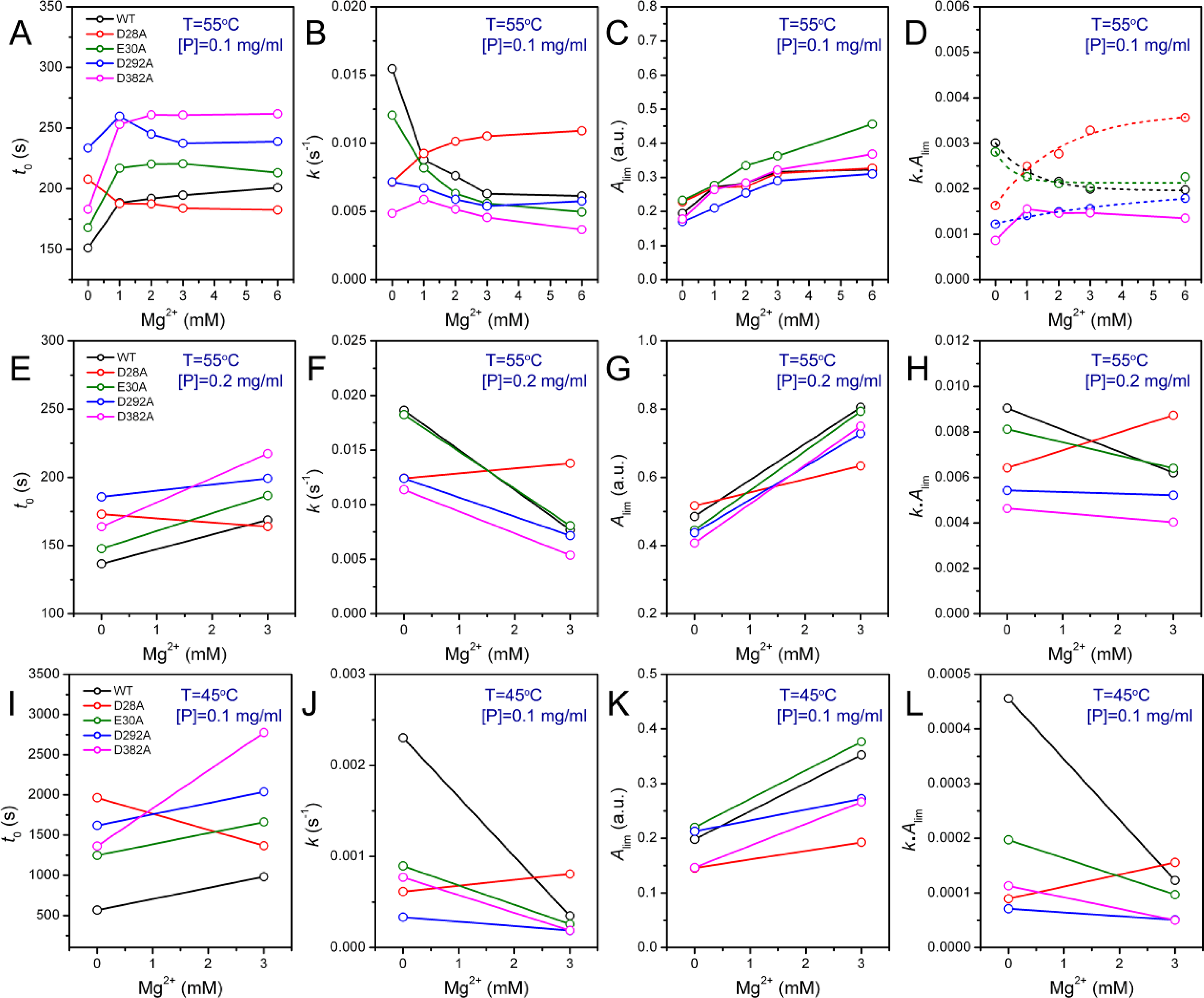
2.3. Effect of Mutations on PARN Thermal Aggregation
The thermal aggregation of PARN was [Mg
2+] dependent, and reached an equilibrium at [Mg
2+] > 3 mM. This suggested that the coordination of Mg
2+ to the active site might be correlated to the effects of Mg
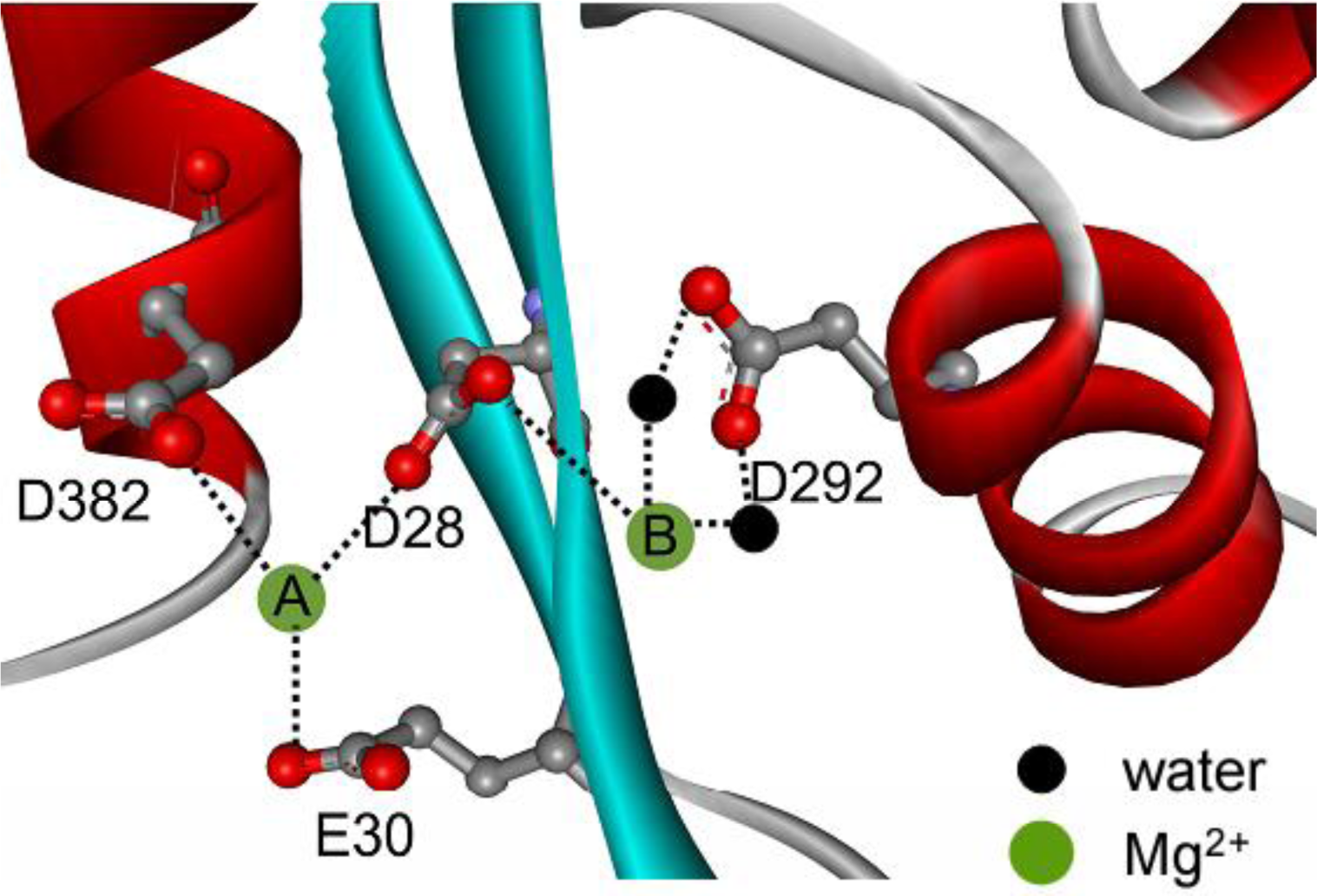
2+ on PARN thermal aggregation. To elucidate this proposal, mutational analysis was carried out with the four conserved acidic residues that directly participated in Mg
2+-binding (
Figure 1). The results indicated that in the absence of Mg
2+, all four mutations significantly decreased PARN aggregation at both 45 °C and 55 °C (
Figures 4 and
6). The aggregation of all mutants exhibited similar temperature-dependence to the WT protein. That is, with the decrease of temperature, the aggregation had a significantly longer
t0, a smaller
k and an almost unchanged
Alim. When protein concentration was increased from 0.1 to 0.2 mg/mL, the changes in
t0 and
k were minor, while an about 2-fold increase in
Alim was observed for all proteins.
As presented in
Figure 6, the addition of Mg
2+ increased the
Alim values of all proteins. However, the Mg
2+ dependence of the other kinetic parameters was dramatically different. Among the four mutants, the E30A mutation resulted in the least changes in the aggregation of PARN under all experimental conditions, and its Mg
2+-dependent property was very similar to the WT protein except that E30A aggregated slower. The most striking observation is that D28A revealed reverse Mg
2+ dependence when compared to that of the WT protein. In the absence of Mg
2+, the E28A mutation decreased PARN aggregation, which is similar to the other three mutants. However, in contrast to the aggregate-inhibition effects of Mg
2+ on the WT and the other three mutants, the addition of Mg
2+ significantly increased PARN aggregation by decreasing
t0 and increasing
k,
Alim and
kAlim. In the presence of 3 mM Mg
2+, the aggregation of D28A was more serious than the WT protein. In most circumstances, D292A and D382A were the two most effective mutations in inhibiting PARN thermal aggregation. The performance of D292A and D382A was between that of D28A and E30A. The Mg
2+ dependence of D382A was close to that of E30A for most cases, while D292A was somewhat more like D28A. It is worth noting that the impact of Mg
2+ on D292A thermal aggregation was weak and was not as obvious as the other three mutants.
2.4. Discussion
The structural stability of multimeric/multi-domain proteins depends not only on the properties of individual domains but also on the domain interactions [
24,
25], and thus protein stability analysis can also provide insights into protein structure and regulation. The full-length PARN is a dimeric enzyme containing three well-defined domains: The nuclease domain, the R3H domain and the RRM [
16]. In addition to the well-folded domains, the C-terminal domain of PARN has recently been proposed to be intrinsically disordered [
26]. The 54 kDa isoform is the N-terminal proteolytic product of the full-length PARN [
23] with the removal of the C-terminal domain and half of the RRM [
20]. The 54 kDa PARN contains two Trp residues: W219 at the R3H domain and W456 at the truncated RRM domain [
20]. Although no Trp residue is located at the catalytic nuclease domain, the significant changes in the Trp fluorescence induced by Mg
2+ coordination (
Figure 2) strongly suggested that the conformation of the other domains was affected by structural changes of the active site. That is, the domain-interaction interface might be close to the active site. This opinion was confirmed by mutational analysis, which indicated that all four mutations at the active site resulted in a dramatic increase in the fluorescence intensity. Indeed, very recently, structural analysis indicated that the RRM interacts with the catalytic domain [
27]. Our results further suggested that Mg
2+ coordinated in the active site not only played an essential role in the two-metal catalysis of PARN, but also induced a large scale conformational change. Although the maximum Trp fluorescence wavelength was not affected by Mg
2+-coordination or mutations, the increase of Trp fluorescence intensity implied that the cofactor or mutation decreased the flexibility around the Trp residues. Meanwhile, a slight increase in the ellipticity was also observed for Mg
2+-coordination and most mutations except D28A. Thus these spectroscopic results suggested that the Mg
2+ coordination or mutations might modulate the repulsion of the negative charges of the four acidic residues in the active site, which further induced large conformational changes of the PARN molecule to a more compact one. Although this conclusion was obtained by studies of the 54 kDa PARN isoform, it could also be true for the 74 kDa full length PARN. In the DEDD nucleases, a fifth residue, which is either His or Tyr, also contributes to the enzymatic activity. PARN belongs to the DEDDh subfamily, and His337 is the fifth residue. However, His337 does not contribute to the coordination of the metal ions, and may not involve in the structural changes induced by the addition of Mg
2+. Moreover, both the addition of Mg
2+ and mutations of the four acidic residues significantly decreased PARN thermal aggregation (
Figure 6), although the active site is buried in the catalytic domain and seems not to participate in PARN aggregation [
28]. Considering that PARN is an allosteric enzyme [
29], it is important to find that the Mg
2+ coordination could induce conformational changes in the other domains of PARN, which might contribute to the allosteric regulation of PARN. This conclusion is quite consistent with the previous observations by Balatsos
et al., which showed that the coordination of Mg
2+ could release the non-competitive nucleotide inhibitors of PARN [
30]. Our findings is also consistent with that previously observed by Dupureur and Dominguez, which showed that the mutations in the active site of
PvuII endonuclease resulted in significant changes in the conformation and stability of the enzyme [
13]. Particularly, they found that three of the mutations led to a 2–5 kcal/mol stabilization of
PvuII endonuclease, implying that the mutants might have a more compact structure.
In most cases, the binding of the divalent metal ion cofactor stabilizes the protein against stresses. Previous research has indicated that Mg
2+ can retard PARN thermal inactivation, but promote PARN aggregation evidenced by the increase of turbidity [
12]. We found that Mg
2+ had dissimilar effects on the kinetic parameters of PARN thermal aggregation. It is worth noting that under the same protein concentration and incubating conditions, the
Alim value reflects the size of the aggregates as proposed previously [
22]. Thus Mg
2+ decreased the rate of the aggregation, but increased the size of aggregates. This unique effect might be caused by the interference of PARN aggregation pathway by Mg
2+. The little change in secondary and tertiary structures [
12] and the first-order kinetics (
Figure 3) suggested that PARN thermal aggregation was dominated by an unfolding-limited aggregation mechanism (native ↔ unfolded/intermediate → aggregate) [
31]. The fact that Mg
2+ elongated
t0 of the WT protein indicated that the cofactor binding stabilized the active site or native structure, and delayed the unfolding process of PARN. Meanwhile, a decrease in
k suggested that the Mg
2+ might also alter the properties of the intermediate state. The Mg
2+-modified intermediates were prone to form larger aggregates when compared to the apo-protein. The underlying molecular mechanism remains unclear. A possible explanation is that Mg
2+ modulated the negative charge repulsion and facilitated the formation of larger aggregates. Actually, electrostatic interactions have been proposed to contribute to protein aggregation for many proteins [
32–
36], and PARN aggregation could also be modulated by electrostatic interactions during chemical denaturant-induced denaturation [
26].
The aggregation of the four mutants showed quite dissimilar Mg
2+-dependent properties, which might be due to their differential roles in Mg
2+ coordination. It has been proposed that in PARN catalysis, metal ion A is coordinated by D28, E30 and D382, while metal ion B binds with D28 and D292 (
Figure 1) [
17,
18]. Obviously, D28 is the most important one in Mg
2+ coordination of PARN, and it has been observed that among the four acidic residues, D28 is crucial to Fe
2+ coordination when substituted by Ala [
17]. Consistent with this observation, we found that the aggregation of D28A showed a reverse Mg
2+ dependent property when compared to the WT PARN, and this property was not observed or not obvious in the other mutants (
Figures 6). The reason why the D28A mutation had the unique property of reversing the action of Mg
2+ is unclear. A shorter
t0 and a larger
k implied that the addition of Mg
2+ altered both the unfolding and the aggregation stage in the unfolding-limited aggregation of D28A. A possible reason is that the mutation disrupted the correct positioning of both metal A and metal B, and led a change in the active site structure. The misfolded active site, as proposed above, resulted in a global change in PARN conformation and facilitated protein aggregation.
If the two metal ions contributed differently to PARN thermal stability, the behavior of D292A should be different from those of and D382A or E30A. In most cases, the Mg
2+-dependence of D292A aggregation was not as obvious as the other mutants. Meanwhile, the E30A mutant behaved just similar to the WT protein, while the property of D382A was between those of D292A and E30A (
Figure 6). Considering that D292 participates the binding of metal B via two water molecules [
17,
18], the significant changes in Mg
2+ dependence by mutation suggested that weakening the binding of metal B resulted in a more serious disruption of the active site structure. Thus the coordination of metal B (
Figure 1) might be more important to the structural stability of PARN. This might also be the reason why the effect of the D28A mutation was not the sum of that of D292A and D382A/E30A since D28 coordinates metal B directly by its side chains. It is worth noting that metal B in enzymes utilizing two-metal-ion catalysis is catalytically equivalent to the single metal ion in enzymes with one-metal-ion catalysis [
4]. It is well-known that the coordination of the metal ion plays a crucial role in the structure and stability of mononuclear enzymes, and the results herein suggested that metal B in dinuclear enzymes may play a similar structural role. Further research in more binuclear enzymes is needed to prove this proposal.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






