Impaired Mitochondrial Respiratory Functions and Oxidative Stress in Streptozotocin-Induced Diabetic Rats

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Induction of Chronic Diabetes
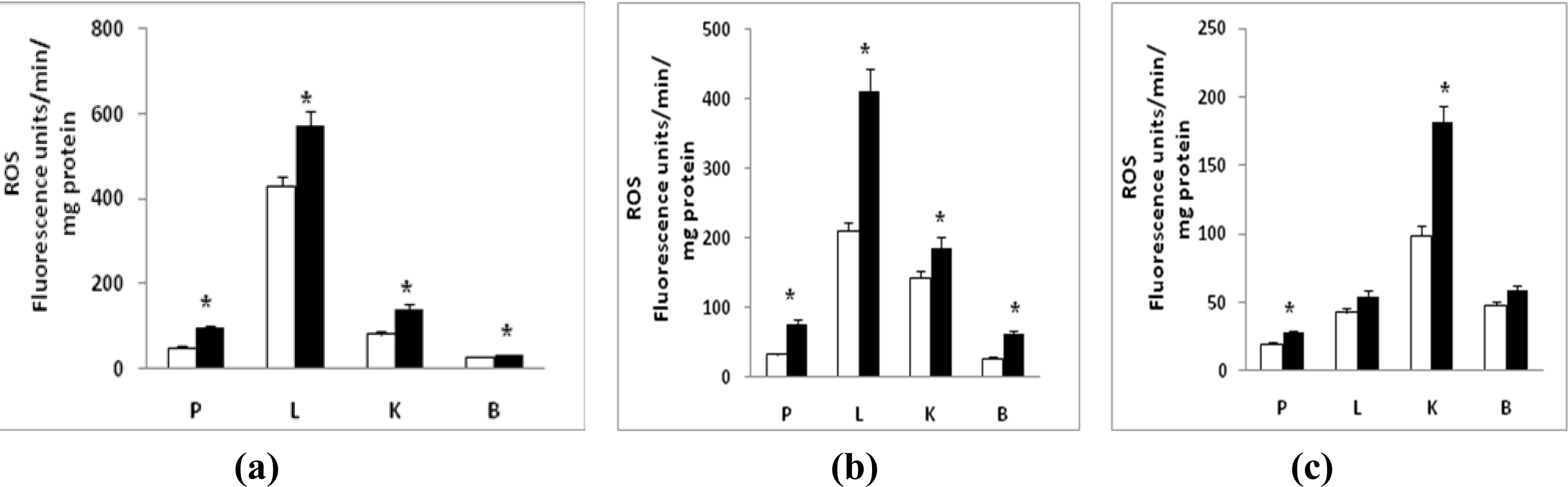
2.2. Sub-Cellular Oxidative Stress
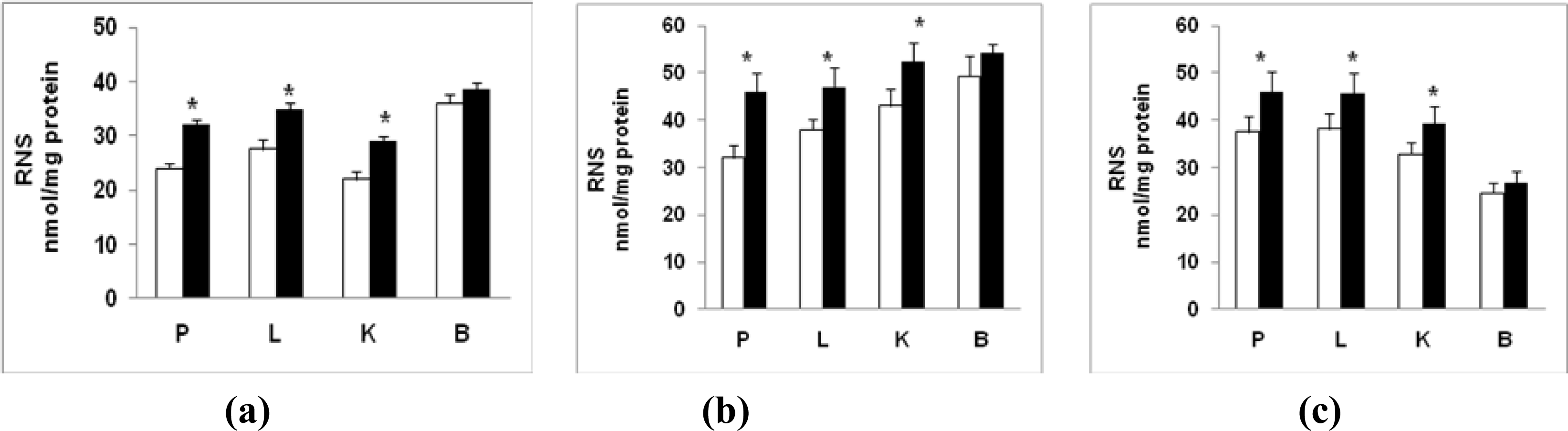
2.3. Nitric Oxide (NO) Production
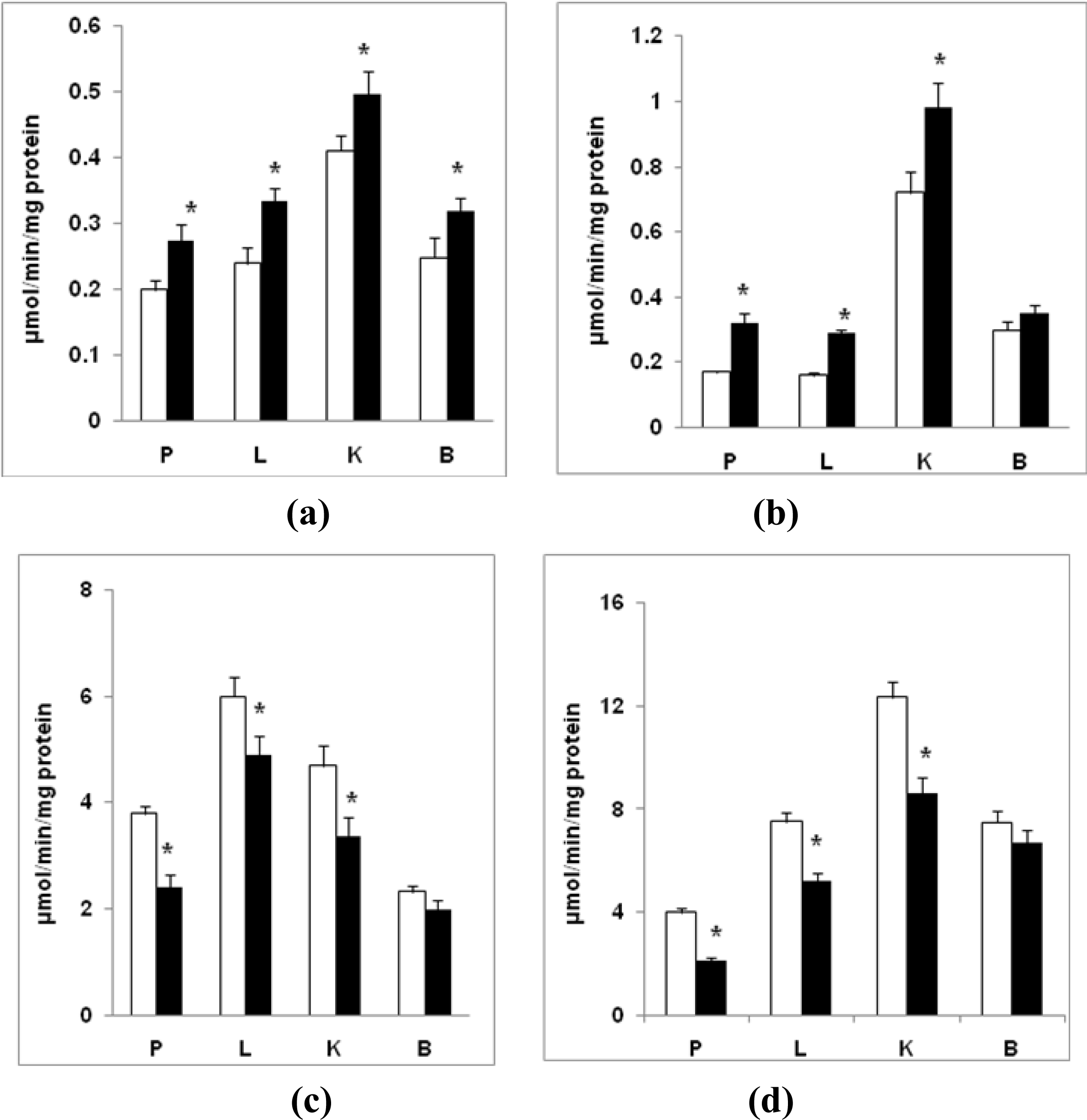
2.4. Oxidative Carbonylation of Proteins
2.5. Respiratory Chain Activities
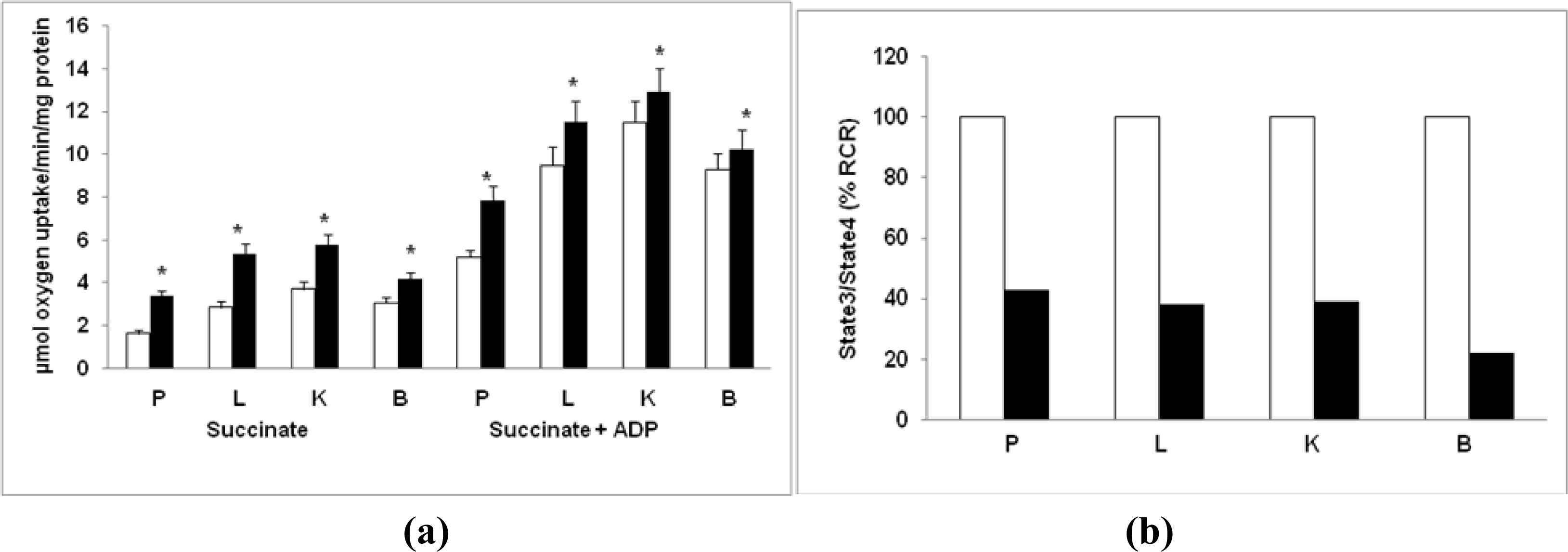
2.6. Mitochondrial O2 Consumption
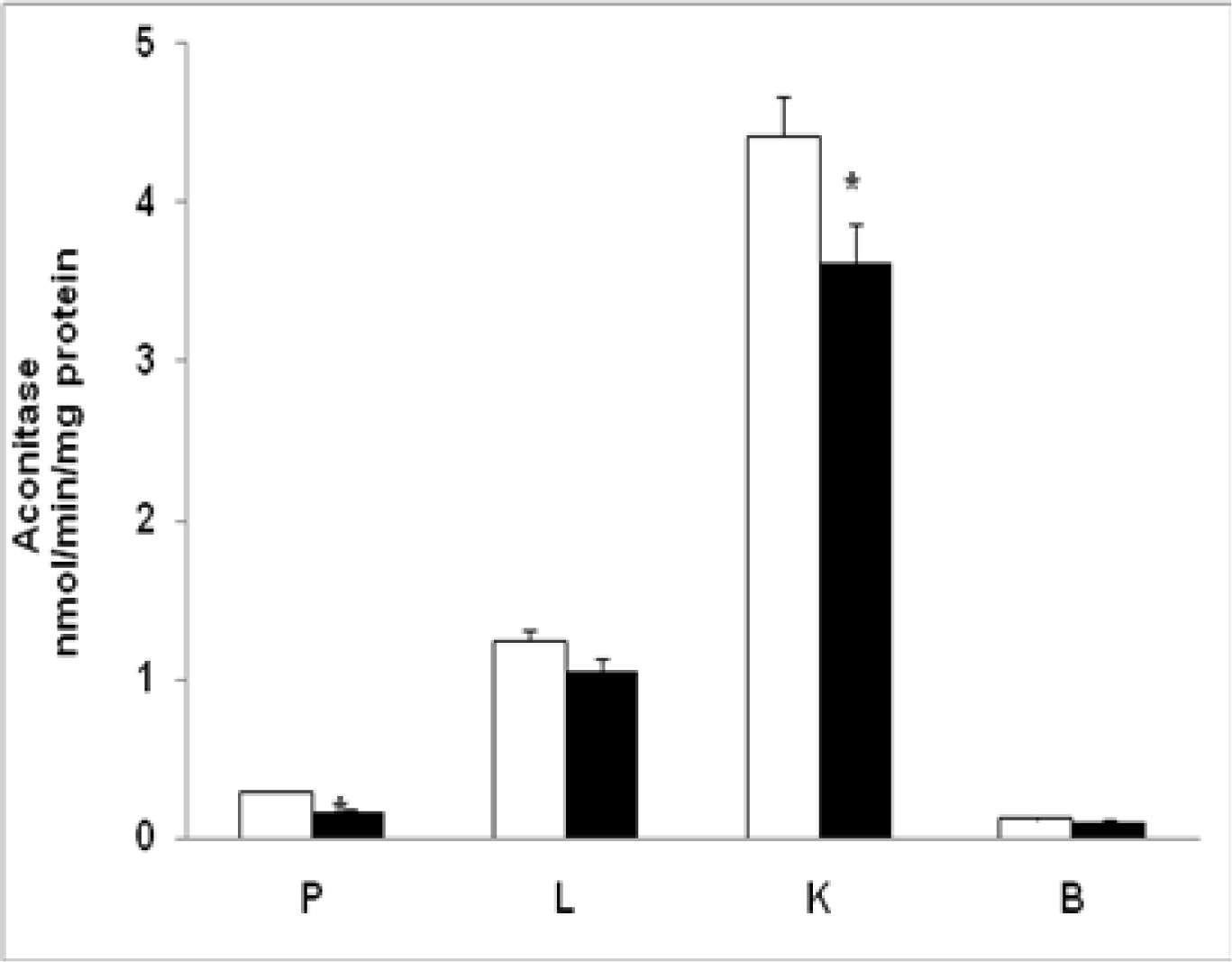
2.7. Mitochondrial Aconitase Activity
2.8. Expression of Complex II, HO-1, Hsp-70 and iNOS in Mitochondria
2.9. Discussion
3. Experimental Section
3.1. Materials
3.2. Treatment of Animals and Sub-Cellular Fractionation
3.3. Assays for Reactive Oxygen and Nitrogen Species (ROS and RNS)
3.4. Assay for Protein Carbonylation
3.5. Measurement of Oxygen Uptake
3.6. Assays for Respiratory Complexes
3.7. SDS-PAGE and Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Abbreviations
| DCFDA | 2′,7′-dichlorofluorescein diacetate; |
| DNPH | 2,4-dinitrophenylhydrazine; |
| GSH | glutathione; |
| NO | nitric oxide; |
| NOS | nitric oxide synthase; |
| ROS | reactive oxygen species; |
| STZ | streptozotocin; |
| SDS-PAGE | sodium dodecylsulfate polyacrylamide gel electrophoresis. |
Acknowledgments
References
- Baynes, J. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar]
- Robertson, RP; Harmon, JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Rad. Biol. Med 2006, 41, 177–184. [Google Scholar]
- Rolo, AP; Palmeira, CM. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharm 2006, 212, 167–178. [Google Scholar]
- Piconi, L; Quagliaro, L; Ceriello, A. Oxidative stress in diabetes. Clin. Chem. Lab. Med 2003, 41, 1144–1149. [Google Scholar]
- Wu, G; Fang, YZ; Yang, S; Lupton, JR; Turner, ND. Glutathione metabolism and its implications for health. J. Nutr 2004, 134, 489–492. [Google Scholar]
- Mastrocola, R; Restivo, F; Vercellinatto, I; Danni, O; Brignardello, E; Aragno, M; Boccuzzi, G. Oxidative and nitrosative stress in brain mitochondria of diabetic rats. J. Endocr 2005, 187, 37–44. [Google Scholar]
- Niedowicz, DM; Daleke, DL. The role of oxidative stress in diabetic complications. Cell Biochem. Biophys 2005, 43, 289–330. [Google Scholar]
- Simmons, RA. Developmental origins of diabetes: The role of oxidative stress. Free Rad. Biol. Med 2006, 40, 917–922. [Google Scholar]
- Sakai, K; Matsumoto, K; Nishikawa, T; Suefuji, M; Nakamaru, K; Hirashima, Y; Kawashima, J; Shirotani, T; Ichinose, K; Brownlee, M; Araki, E. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem. Biophys. Res. Comm 2003, 300, 216–222. [Google Scholar]
- Luft, R; Landau, BR. Mitochondrial medicine. J. Int. Med 1995, 238, 405–421. [Google Scholar]
- Kaneto, H; Kajimoto, Y; Miyagawa, J; Matsuoka, T; Fujitani, Y; Umayahara, Y; Hanafusa, T; Matsuzawa, Y; Yamasaki, Y; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar]
- Yu, T; Robotham, JL; Yoon, Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. PNAS (USA) 2006, 103, 2653–2658. [Google Scholar]
- Kucharska, J; Braunova, Z; Ulicna, O; Zlatos, L; Gvozdjakova, A. Deficit of coenzyme Q in heart and liver mitochondria of rats with streptozotocin-induced diabetes. Phys. Res 2000, 49, 411–418. [Google Scholar]
- Wallace, DC. The mitochondrial genome in human adaptive radiation and disease: On the road to therapeutics and performance enhancement. Gene 2005, 354, 169–180. [Google Scholar]
- Sanz, A; Caro, P; Sanchez, JG; Barja, G. Effect of lipid restriction on mitochondrial free radical production and oxidative DNA damage. Ann. N.Y. Acad. Sci 2006, 1067, 200–209. [Google Scholar]
- Vergani, L; Floreani, M; Russell, A; Ceccon, M; Napoli, E; Cabrelle, A; Valente, L; Bragantini, F; Leger, B; Dabbeni-Sala, F. Antioxidant defences and homeostasis of reactive oxygen species in different human mitochondrial DNA-depleted cell lines. Eur. J. Biochem 2004, 271, 3646–3656. [Google Scholar]
- Hunzinger, C; Wozny, W; Schwall, GP; Poznanovic, S; Stegmann, W; Zengerling, H; Schoepf, R; Groebe, K; Cahill, MA; Osiewacz, HD; Jagemann, N; Bloch, M; Dencher, NA; Krause, F; Schrattenholz, A. Comparative profiling of the mammalian mitochondrial proteome: Multiple aconitase-2 isoforms including N-formylkynurenine modifications as part of a protein biomarker signature for reactive oxidative species. J. Proteome Res 2006, 5, 625–633. [Google Scholar]
- Santos, DL; Palmeira, CM; Seica, R; Dias, J; Mesquita, J; Moreno, AJ; Santos, MS. Diabetes and mitochondrial oxidative stress: A study using heart mitochondria from the diabetic Goto-Kakizaki rat. Mol. Cell. Biochem 2003, 246, 163–170. [Google Scholar]
- Duchen, MR. Mitochondria in health and disease: Perspectives on a new mitochondrial biology. Mol. Asp. Med 2004, 25, 365–451. [Google Scholar]
- Green, K; Brand, MD; Murphy, MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004, 53, S110–S118. [Google Scholar]
- Michelakis, ED; Hampl, V; Nsair, A; Wu, X; Harry, G; Haromy, A; Gurtu, R; Archer, SL. Diversity in mitochondrial function explains differences in vascular oxygen sensing. Circ. Res 2002, 2890, 1307–1315. [Google Scholar]
- Demin, OV; Kholodenko, BN; Skulachev, VP. A model of O•2− generation in the complex III of the electron transport chain. Mol. Cell. Biochem 1998, 184, 21–33. [Google Scholar]
- Raza, H; Prabu, SK; Robin, MA; Avadhani, NG. Elevated mitochondrial cytochrome P450 2E1 and glutathione S-transferase A4–4 in streptozotocin-induced diabetic rats: Tissue-specific variations and roles in oxidative stress. Diabetes 2004, 53, 185–194. [Google Scholar]
- Prabu, SK; Anandatheerthavarada, HK; Raza, H; Srinivasan, S; Spear, JF; Avadhani, NG. Protein kinase A-mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia-related injury. J. Biol. Chem 2006, 281, 2061–2070. [Google Scholar]
- Gardner, PR. Aconitase: Sensitive target and measure of superoxide. Meth. Enzymol 2002, 349, 9–23. [Google Scholar]
- Raza, H; John, A. Glutathione metabolism and oxidative stress in neonatal rat tissues from streptozotocin-induced diabetic mothers. Diab. Metab. Res. Rev 2004, 20, 72–78. [Google Scholar]
- Raza, H; Robin, MA; Fang, JK; Avadhani, NG. Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. Biochem. J 2002, 366, 45–55. [Google Scholar]
- Miwa, S; Brand, MD. Mitochondrial matrix reactive oxygen species production is very sensitive to mild uncoupling. Biochem. Soc. Transac 2003, 31, 1300–1301. [Google Scholar]
- Chen, Q; Vazquez, EJ; Moghaddas, S; Hoppel, CL; Lesnefsky, EJ. Production of reactive oxygen species by mitochondria. J. Biol. Chem 2003, 278, 36027–36031. [Google Scholar]
- Han, D; Canali, R; Rettori, D; Kaplowitz, N. Effect of glutathione depletion on sites and topology of superoxide and hydrogen peroxide production in mitochondria. Mol. Pharm 2003, 64, 1136–1144. [Google Scholar]
- Muller, FL; Liu, Y; van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem 2004, 279, 49064–49073. [Google Scholar]
- Yoon, YS; Lee, JH; Hwang, SC; Choi, KS; Yoon, G. TGF beta1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar]
- Lluis, JM; Morales, A; Blasco, C; Colell, A; Mari, M; Garcia-Ruiz, C; Fernandez-Checa, JC. Critical role of mitochondrial glutathione in the survival of hepatocytes during hypoxia. J. Biol. Chem 2005, 280, 3224–3232. [Google Scholar]
- Guzy, RD; Schumacker, PT. Oxygen sensing by mitochondria at complex III: The paradox of increased ROS during hypoxia. Exp. Physiol 2006, 91, 807–819. [Google Scholar]
- Grivennikova, VG; Vinogradov, AD. Generation of superoxide by the mitochondrial Complex I. Biochim. Biophys. Acta 2006, 1757, 553–561. [Google Scholar]
- Katyare, SS; Satav, JG. Effect of streptozotocin-induced diabetes on oxidative energy metabolism in rat kidney mitochondria. A comparative study of early and late effects. Diab. Obes. Metab 2005, 7, 555–562. [Google Scholar]
- Schild, L; Reinheckel, T; Reiser, M; Horn, TF; Wolf, G; Augustin, W. Nitric oxide produced in rat liver mitochondria causes oxidative stress and impairment of respiration after transient hypoxia. FASEB J 2003, 17, 2194–2201. [Google Scholar]
- Shiva, S; Oh, JY; Landar, AL; Ulasova, E; Venkatraman, A; Bailey, SM; Darley-Usmar, VM. Nitroxia: The pathological consequence of dysfunction in the nitric oxide-cytochrome c oxidase signaling pathway. Free Rad. Biol. Med 2005, 38, 297–306. [Google Scholar]
- McCabe, C; Samali, A; O’brien, T. Beta cell cytoprotective strategies: Establishing the relative roles for iNOS and ROS. Biochem. Biophys. Res. Comm 2006, 342, 1240–1248. [Google Scholar]
- Choksi, KB; Boylston, WH; Rabek, JP; Widger, WR; Papaconstantinou, J. Oxidatively damaged proteins of heart mitochondrial electron transport complexes. Biochim. Biophys. Acta 2004, 1688, 95–101. [Google Scholar]
- Chen, J; Henderson, GI; Freeman, GL. Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. J. Mol. Cell. Card 2001, 33, 1919–1927. [Google Scholar]
- Turella, P; Pedersen, JZ; Caccuri, AM; De Maria, F; Mastroberardino, P; Lo Bello, M; Federici, G; Ricci, G. Glutathione transferase superfamily behaves like storage proteins for dinitrosyl-diglutathionyl-iron complex in heterogeneous systems. J. Biol. Chem 2003, 278, 42294–42299. [Google Scholar]
- Shiva, S; Crawford, JH; Ramachandran, A; Ceaser, EK; Hillson, T; Brookes, PS; Patel, RP; Darley-Usmar, VM. Mechanisms of the interaction of nitroxyl with mitochondria. Biochem. J 2004, 379, 359–366. [Google Scholar]
- Lowry, OH; Rosebrough, NJ; Farr, AL; Randall, RJ. Protein measurement with Folin Phenol reagent. J. Biol. Chem 1951, 193, 265–275. [Google Scholar]
- Levine, RL; Williams, JA; Stadtman, ER; Shacter, E. Carbonyl assays for determination of oxidatively modified proteins. Meth. Enzymol 1994, 233, 346–357. [Google Scholar]
- Laemmli, UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Towbin, H; Staehelin, T; Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 1979, 76, 4350–4354. [Google Scholar]







© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Raza, H.; Prabu, S.K.; John, A.; Avadhani, N.G. Impaired Mitochondrial Respiratory Functions and Oxidative Stress in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2011, 12, 3133-3147. https://doi.org/10.3390/ijms12053133
Raza H, Prabu SK, John A, Avadhani NG. Impaired Mitochondrial Respiratory Functions and Oxidative Stress in Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences. 2011; 12(5):3133-3147. https://doi.org/10.3390/ijms12053133
Chicago/Turabian StyleRaza, Haider, Subbuswamy K. Prabu, Annie John, and Narayan G. Avadhani. 2011. "Impaired Mitochondrial Respiratory Functions and Oxidative Stress in Streptozotocin-Induced Diabetic Rats" International Journal of Molecular Sciences 12, no. 5: 3133-3147. https://doi.org/10.3390/ijms12053133