An Optimized Protocol for DNA Extraction from Wheat Seeds and Loop-Mediated Isothermal Amplification (LAMP) to Detect Fusarium graminearum Contamination of Wheat Grain

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Germination of Seeds
2.3. Wheat Seedling Inoculation
2.4. Fungal Cultures
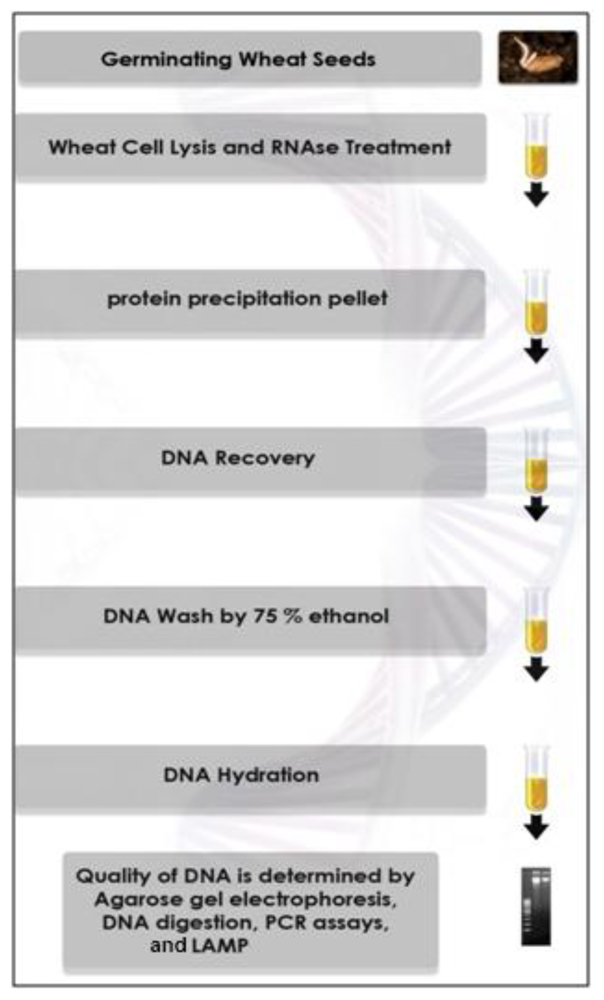
2.5. DNA Extraction
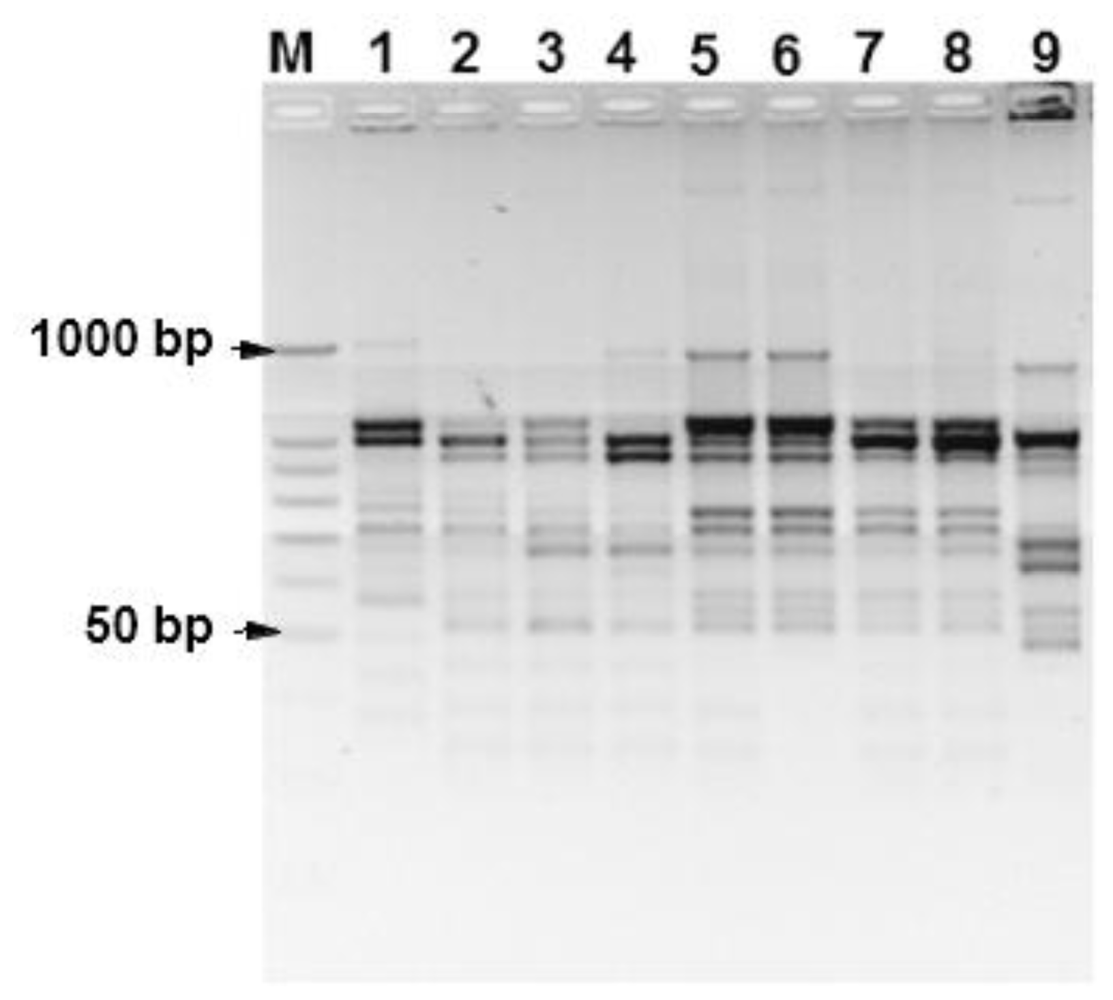
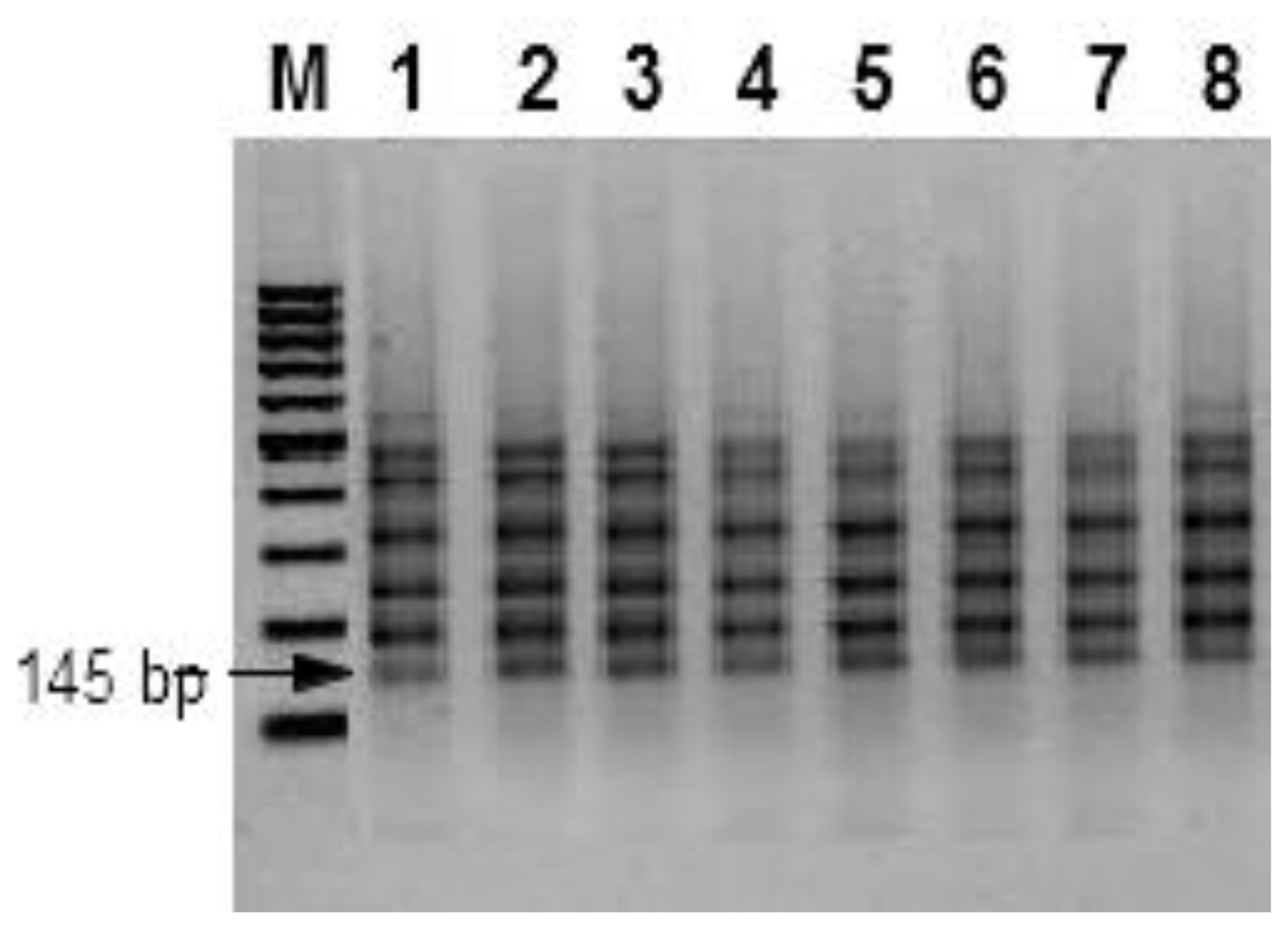
2.6. Inter-Simple Sequence Repeats (ISSRs)
2.7. Trichodiene Synthase (Tri5) Gene Assay
2.8. LAMP Reaction
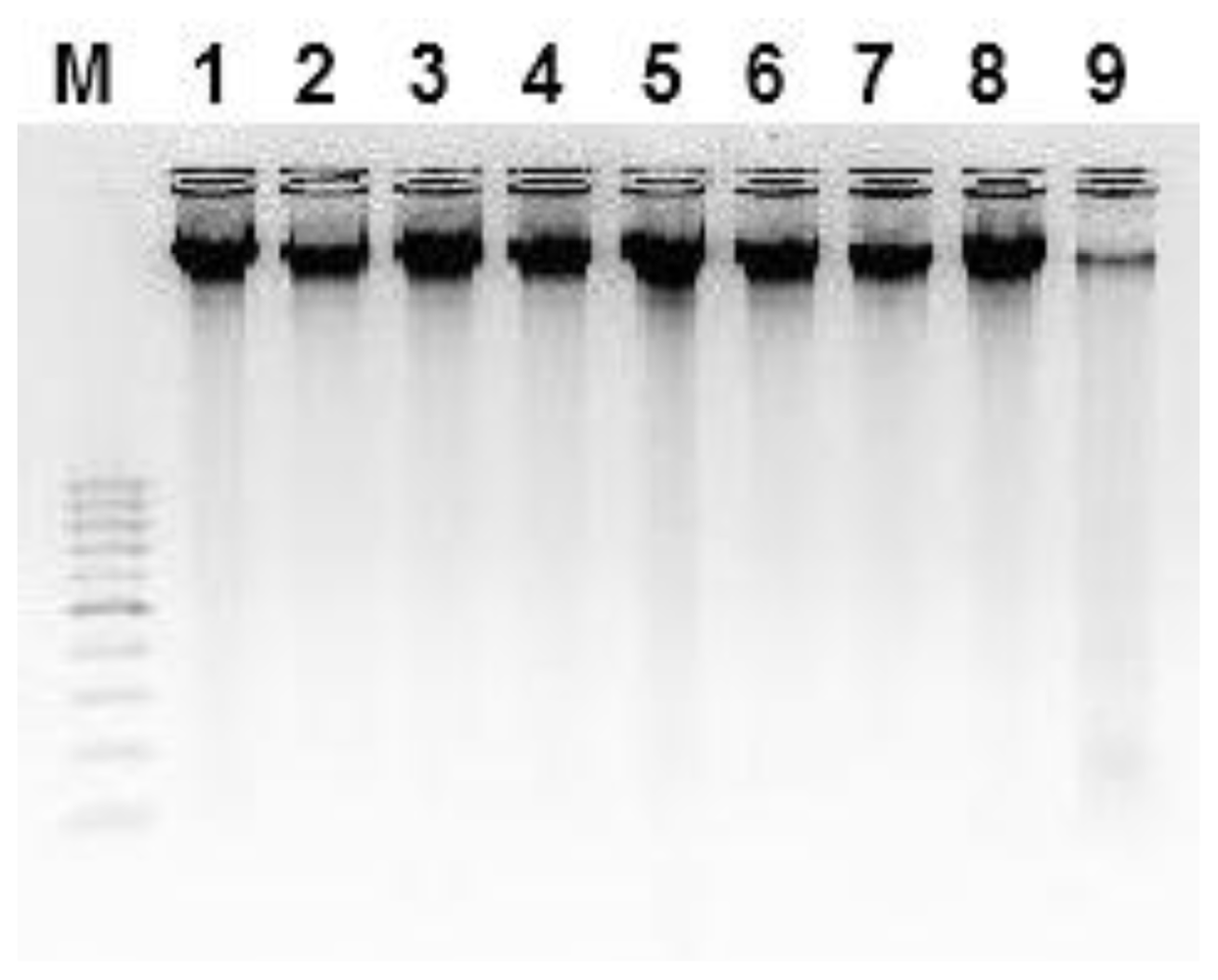
3. Results
4. Discussion
5. Conclusions
Acknowledgements
References
- Sarwat, M; Negi, MS; Lakshmikumaran, M; Tyagi, AK; Das, S; Srivastava, PS. A standardized protocol for genomic DNA isolation from Terminalia arjuna for genetic diversity analysis. Electron. J. Biotechnol 2006, 9, 86–91. [Google Scholar]
- Braid, MD; Daniels, LM; Kitts, CL. Removal of PCR inhibitors from soil DNA by chemical flocculation. J. Microbiol. Methods 2003, 52, 389. [Google Scholar]
- Chungwongse, J; Martin, GB; Tanksley, SD. Pregermination genotypic screening using PCR amplification of half-seeds. Theor. Appl. Genet 1993, 86, 694–698. [Google Scholar]
- Kang, HW; Cho, YG; Yoon, UH; Eun, MY. A rapid DNA extraction method for RFLP and PCR analysis from single dry seed. Plant Mol. Biol. Report 1998, 16, 1–9. [Google Scholar]
- Desjardins, AE. Fusarium Mycotoxins Chemistry, Genetics and Biology; APS Press: St. Paul, MN, USA, 2006; pp. 145–194. [Google Scholar]
- Niessen, L. PCR-based detection and quantification of mycotoxin-producing fungi. Adv. Food Nutr. Res 2008, 54, 81–138. [Google Scholar]
- Mulé, G; Gonzalez-Jaen, MT; Hornok, L; Nicholson, P; Waalwijk, C. Advances in molecular diagnosis of toxigenic Fusarium species: A review. Food Addit. Contam 2005, 22, 316–323. [Google Scholar]
- Schnerr, H; Vogel, RF; Niessen, L. Correlation between DNA of tricothecene-producing Fusarium species and deoxynivalenol concentrations in wheat-samples. Lett. Appl. Microbiol 2002, 35, 121–125. [Google Scholar]
- Parida, M; Sannarangaiah, S; Dash, PK; Rao, PV; Morita, K. Loop mediated isothermal amplification (LAMP): A new generation of innovative gene amplification technique; perspectives in clinical diagnosis of infectious diseases. Rev. Med. Virol 2008, 18, 407–421. [Google Scholar]
- Endo, S; Komori, T; Ricci, G; Sano, A; Yokoyama, K; Ohori, A; Kamai, K; Franco, M; Miyaji, M; Nishimura, K. Detection of gp43 of Paracoccidioides brasiliensis by the loop-mediated isothermal amplification (LAMP) method. FEMS Microbiol. Lett 2004, 234, 93–97. [Google Scholar]
- Ohori, A; Endo, S; Yokoyama, K; Yarita, K; Yamaguchi, M; Kamai, K; Miyaji, M; Nishimura, K. Rapid identification of Ochroconis gallopava by a loop-mediated isothermal amplification (LAMP) method. Vet. Microbiol 2006, 114, 359–365. [Google Scholar]
- Uemura, N; Makimura, K; Onozaki, M; Otsuka, Y; Shibuya, Y; Yazaki, H; Kikuchi, Y; Abe, S; Kudoh, S. Development of a loop-mediated isothermal amplification method for diagnosis of Pneumocystis pneumonia. J. Medic. Microbiol 2008, 57, 50–57. [Google Scholar]
- Matsuzawa, T; Tanaka, R; Horie, Y; Gonoi, T; Yaguchi, T. Development of rapid and specific molecular discrimination methods for pathogenic Emericella species. Jpn. J. Med. Mycol 2010, 51, 109–116. [Google Scholar]
- Sun, J; Najafzadeh, MJ; Vicente, V; Xi, L; de Hoog, GS. Rapid detection of pathogenic fungi using loop-mediated isothermal amplification, exemplified by Fonsecaea agents of chromoblastomycosis. J. Microbiol. Methods 2010, 80, 19–24. [Google Scholar]
- Tomlinson, JA; Barker, I; Boonham, N. Faster, simpler, more-specific methods for improved molecular detection of Phytophthora ramorum in the field. Appl. Environ. Microbiol 2007, 73, 4040–4047. [Google Scholar]
- Gadkar, V; Rillig, MC. Evaluation of loop-mediated isothermal amplification (LAMP) to rapidly detect arbuscular mycorrhizal fungi. Soil Biol. Biochem 2008, 40, 540–543. [Google Scholar]
- Hayashi, N; Arai, R; Tada, S; Taguchi, H; Ogawa, Y. Detection and identification of Brettanomyces/Dekkera sp. yeasts with a loop-mediated isothermal amlificatioin method. Food Microbiol 2007, 24, 778–785. [Google Scholar]
- Notomi, T; Okayama, H; Masubuchi, H; Yonekawa, T; Watanabe, K; Amino, N; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res 2000, 28, e63. [Google Scholar]
- Niessen, L; Vogel, RF. Detection of Fusarium graminearum DNA using a loop-mediated isothermal amplification (LAMP) assay. Int. J. Food Microbiol 2010, 140, 183–191. [Google Scholar]
- Niessen, L; Vogel, RF. Group specific PCR-detection of potential trichochecene producing Fusarium-species in pure cultures and cereal samples. Syst. Appl. Microbiol 1998, 21, 618–621. [Google Scholar]
- Nagaoka, T; Ogihara, Y. Applicability of inter-simple sequence repeat polymorphisms in wheat for use as DNA markers in comparison to RFLP and RAPD markers. Theor. Appl. Genet 1997, 94, 597–602. [Google Scholar]
- Hilton, AJ; Jenkinson, P; Hollins, TW; Parry, DW. Relationship between cultivar height and severity of Fusarium ear blight in wheat. Plant Pathol 1999, 48, 202–208. [Google Scholar]
- Nirenberg, H. A simplified method for identifying Fusarium species occurring in wheat. Can. J. Bot 1981, 59, 1599–1609. [Google Scholar]
- Moslem, MA; Bahkali, AH; Abd-Elsalam, KA; Wit, PJGM. An efficient method for DNA extraction from Cladosporioid fungi. Genet. Mol. Res 2010, 9, 2283–2291. [Google Scholar]
- Tomita, N; Mori, Y; Kanda, H; Notomi, T. Loop-mediated isothermal amplification (LAMP) of gene sequences and simple visual detection of products. Nat. Protoc 2008, 3, 877–882. [Google Scholar]
- Abd-Elsalam, KA; Asran-Amal, A; El-Samawaty, A. Isolation of high quality DNA from cotton and its fungal pathogens. J. Plant Dis. Prot 2007, 114, 113–116. [Google Scholar]
- Knoll, S; Mulfinger, S; Niessen, L; Vogel, RF. Rapid preparation of Fusarium DNA from cereals for diagnostic PCR using sonification and an extraction kit. Plant Pathol 2002, 51, 728–734. [Google Scholar]
- Nicolaisen, M; Supronienė, S; Nielsen, KL; Lazzaro, I; Spliid, NH; Justesen, AF. Real-time PCR for quantification of eleven individual Fusarium species in cereals. J. Microbiol. Methods 2009, 76, 234–240. [Google Scholar]
- Zhang, J; Stewart, JM. Economical and rapid method for extracting cotton genomic DNA. J. Cotton Sci 2000, 4, 193–201. [Google Scholar]
- Dean, TR; Roop, B; Betancourt, D; Menetrez, MY. A simple multiplex polymerase chain reaction assay for the identification of four environmentally relevant fungal contaminants. J. Microbiol. Methods 2005, 61, 9–16. [Google Scholar]
- Deshmukh, VP; Thakare, PV; Chaudhari, US; Gawande, PA. A simple method for isolation of genomic DNA from fresh and dry leaves of Terminalia arjuna (Roxb.) Wight and Argot. Electron. J. Biotechnol 2007, 10, 468–472. [Google Scholar]
- Ribeiro, RA; Lovato, MB. Comparative analysis of different DNA extraction protocols in fresh and herbarium specimens of the genus Dalbergia. Genet. Mol. Res 2007, 6, 173–187. [Google Scholar]
- Zidani, S; Ferchichi, A; Chaieb, M. Genomic DNA extraction method from earl millet (Pennisetum glaucum) leaves. Afr. J. Biotechnol 2005, 8, 862–866. [Google Scholar]
- Weising, K; Nybom, H; Wolff, K; Kahl, G. DNA Fingerprinting in Plants: Principles, Methods and Applications, 2nd ed; CRC Press: Boca Raton, FL, USA, 2005; pp. 106–107. [Google Scholar]
- Christopher, M; Cordeiro, G; Waters, D; Henry, R. Marker Assisted Selection in Rice Improvement. In A Report for the Rural Industries Research and Development Corporation; RIRDC Publication No. 04/011; RIRDC: Kingston, Australia, 2004. [Google Scholar]
- Nagamine, K; Watanabe, K; Ohtsuka, K; Hase, T; Notomi, T. Loop-meidated isothermal amplification reaction using a non denatured template. Clin. Chem 2001, 9, 1742–1743. [Google Scholar]
- Hill, J; Beriwal, S; Chandra, I; Paul, VK; Kapil, A; Singh, T; Wadowsky, RM; Singh, V; Goyal, A; Jahnukainen, T; et al. Loop-mediated isothermal amplification assay for rapid detection of common strains of Escherichia coli. J. Clin. Microbiol 2008, 46, 2800–2804. [Google Scholar]
- Poon, LL; Wong, BW; Ma, EH; Chan, KH; Chow, LM; Abeyewickreme, W; Tangpukdee, N; Yuen, KY; Guan, Y; Looareesuwan, S; Peiris, JS. Sensitive and inexpensive molecular test for falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin. Chem 2006, 52, 303–306. [Google Scholar]
- Qiao, YM; Guo, YC; Zhang, XE; Zhou, YF; Zhang, ZP; Wei, HP; Yang, RF; Wang, DB. Loop-mediated isothermal amplification for rapid detection of Bacillus anthracis spores. Biotechnol. Lett 2007, 29, 1939–1946. [Google Scholar]
- Boehme, CC; Nabeta, P; Henastroza, G; Rubhana, R; Rahim, Z; Gerhardt, M; Sanga, E; Hoelscher, M; Notomi, T; Hase, T; Perkins, MD. Operational feasibility of using loop-mediated isothermal amplification (LAMP) for the diagnosis of pulmonary TB in microscopy centres of developing countries. J. Clin. Microbiol 2007, 45, 1936–1940. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungal Species | Reference Strain | tri5 Gene | LAMP GaoA ID4 |
|---|---|---|---|
| F. avenaceum | DSMZ 62785 | − | − |
| F. avenaceum | TMW 4.0140 | − | − |
| F. cerealis | DSMZ 8704 | + | − |
| F. chlamydosporum | DSMZ 62169 | + | − |
| F. chlamydosporum | PPathRI 0222 | + | − |
| F. concolor | DSMZ 62179 | + | − |
| F. culmorum | DSMZ 1094 | + | − |
| F. graminearum | PPathRI 0555 | + | + |
| F. graminearum | DSMZ 1096 | + | + |
| F. graminearum | DSMZ 893 | + | + |
| F. graminearum | TMW 4.0208 | + | + |
| F. langsethiae | TMW 4.0072 | + | − |
| F. incarnatum | DSMZ 62403 | + | − |
| F. proliferatum | DSMZ 62376 | + | − |
| F. verticillioides | DSMZ 62452 | + | − |
| F. oxysporum | PPathRI 0003 | − | − |
| F. oxysporum f. sp. vasinfectum | PPathRI 0421 | − | − |
| Alternaria alternata | PPathRI 0500 | − | − |
| Aspergillus flavus | PPathRI 0111 | − | − |
| Macrophomina phaseolina | PPathRI 0342 | − | − |
| Rhizoctonia solani | PPathRI 0471 | − | − |
| Primer Code | Primer Sequence | Concentrations |
|---|---|---|
| ISSR W8 | 5′-CTC TCT CTC TCT CTC TCT-3′ | 20 pmol |
| Tox5-1 | 5′-GCT GCT CAT CAC TTT GCT CAG-3′ | 20 pmol |
| Tox5-2 | 5′CTG ATC TGG TCA CGC TCA TC-3′ | 20 pmol |
| FIP-gaoA ID4 | 5′-CGC AAG TGA CGG CCC AGT TGC TTC GAG CCT CAG CAC CTA-3′ | 1.6 mM |
| BIP-gaoA ID4 | 5′-TGC AAC AAG GCC ATT GAT GGC CGT TGG CGC CAT AGA ATG T-3′ | 1.6 mM |
| F3-gaoA ID4 | 5′-AGG GAG TCT TCA GTT CCT GA-3′ | 0.2 mM |
| B3-gaoA ID4 | 5′-GTG AGG GGG CTT TGG ATC-3′ | 0.2 mM |
| LoopF-gaoA ID4 | 5′-GTT GCG AGA AAT GGC GCT TCC G-3′ | 0.8 mM |
| LoopB-gaoA ID4 | 5′-ACA AGG ATA CCT TTT GGC AC-3′ | 0.8 mM |
© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Abd-Elsalam, K.; Bahkali, A.; Moslem, M.; Amin, O.E.; Niessen, L. An Optimized Protocol for DNA Extraction from Wheat Seeds and Loop-Mediated Isothermal Amplification (LAMP) to Detect Fusarium graminearum Contamination of Wheat Grain. Int. J. Mol. Sci. 2011, 12, 3459-3472. https://doi.org/10.3390/ijms12063459
Abd-Elsalam K, Bahkali A, Moslem M, Amin OE, Niessen L. An Optimized Protocol for DNA Extraction from Wheat Seeds and Loop-Mediated Isothermal Amplification (LAMP) to Detect Fusarium graminearum Contamination of Wheat Grain. International Journal of Molecular Sciences. 2011; 12(6):3459-3472. https://doi.org/10.3390/ijms12063459
Chicago/Turabian StyleAbd-Elsalam, Kamel, Ali Bahkali, Mohamed Moslem, Osama E. Amin, and Ludwig Niessen. 2011. "An Optimized Protocol for DNA Extraction from Wheat Seeds and Loop-Mediated Isothermal Amplification (LAMP) to Detect Fusarium graminearum Contamination of Wheat Grain" International Journal of Molecular Sciences 12, no. 6: 3459-3472. https://doi.org/10.3390/ijms12063459